The FDA's January 2026 revised clinical decision support guidance matters for AI-enabled CDS, but it is not an AI rulebook. That distinction matters. The guidance creates real room for some software developers to argue that their CDS tools should remain outside active device oversight, or at least within FDA enforcement discretion. It also leaves untouched the questions that have become hardest in practice: generative AI, LLM-enabled clinical advice, consumer-facing health chatbots, and hospital-built predictive models.
Read closely, the update is not deregulatory theater. FDA changed operative positions in the CDS framework, including how it treats a single clinically appropriate recommendation, how it handles time-critical use, and what kind of transparency may be enough for independent clinician review.[1] But anyone trying to turn the document into a broad AI compliance answer will run out of text quickly.

The Change That Will Matter First: One Recommendation Can Be Enough
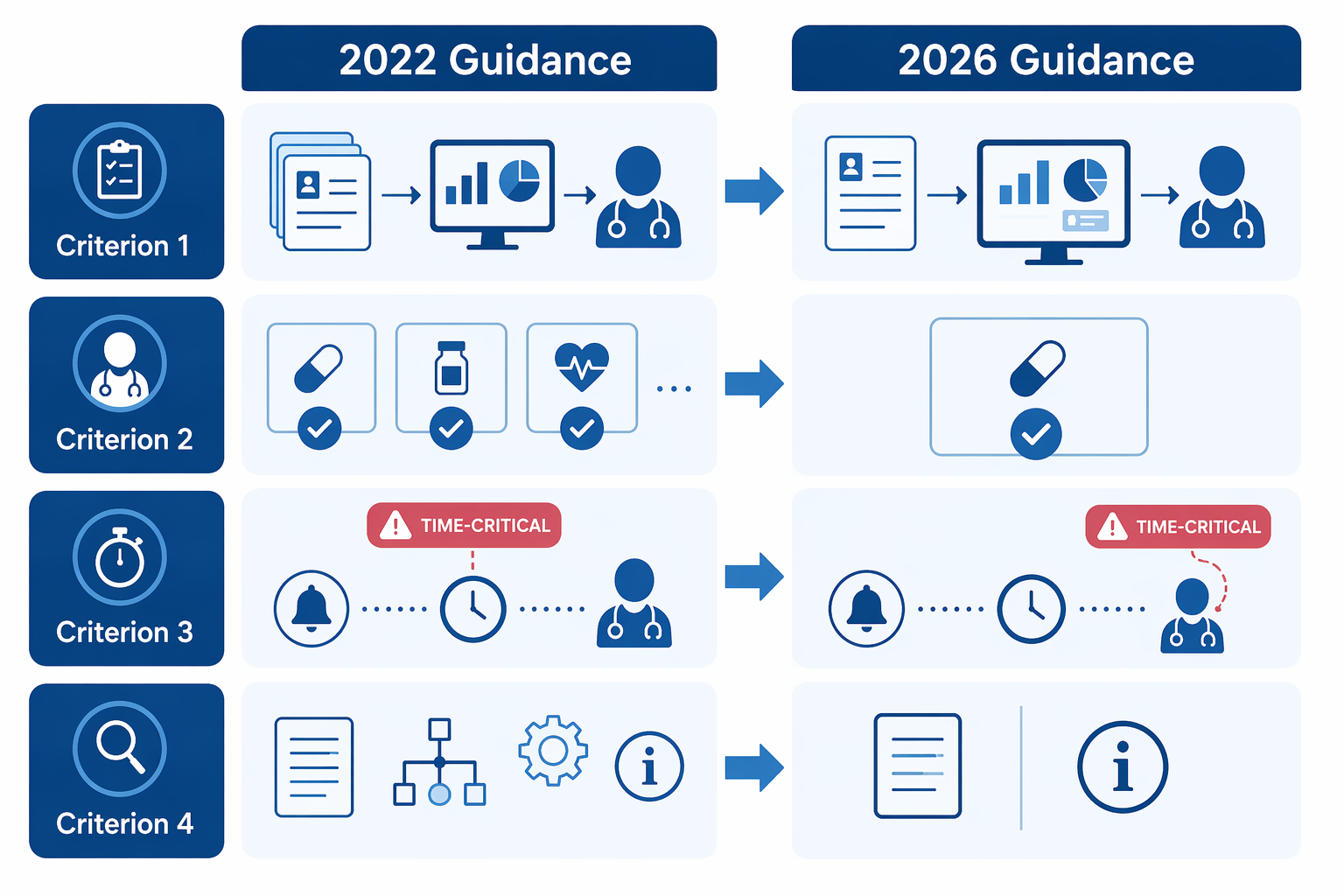
The most commercially important change is FDA's new willingness to exercise enforcement discretion for certain CDS software that provides a single output when only one option is clinically appropriate, so long as the other non-device criteria are met.[1] That is a material shift from the 2022 posture, which pushed developers toward presenting a list of options rather than one recommendation.[2][3][4]
For AI-enabled CDS, this is the part that changes design meetings. Many useful tools do not naturally produce a menu. A system that flags a medication interaction, proposes a follow-up action tied to a specific guideline, or drafts a documentation summary may have one clinically sensible next step rather than three polite alternatives. The older multi-option framing could make such tools look more device-like simply because they were direct.
The 2026 guidance does not say that every single-output AI recommendation is now low risk. The opening is narrower: the output must fit within the CDS framework, the recommendation must be clinically appropriate in context, and the user still must be able to independently review the basis for the output.[1] A developer cannot bury the method, show a confident answer, and call the clinician's signature independent review.
| Issue | 2026 practical effect | Why it matters for AI CDS |
|---|---|---|
| Single clinically appropriate recommendation | FDA may exercise enforcement discretion if the other non-device criteria are met | Some AI-enabled tools no longer need to force artificial lists of alternatives |
| Time-critical decision-making | The concern is no longer an automatic Criterion 3 exclusion, but it remains relevant under Criterion 4 | Fast clinical contexts still face a hard independent-review question |
| Transparency under Criterion 4 | The guidance emphasizes usable information rather than exhaustive technical detail | Developers may have more room to design clinician-facing explanations, but not to omit the basis for review |
| Clinical documentation tools | New examples show some documentation-adjacent tools may fall within enforcement discretion | AI summarization tied to clinical findings gets a clearer, though still bounded, path |
That is relief, not amnesty. The difference will show up in product documentation. A compliance file for a conventional rules engine may point to source guidelines, input fields, and logic tables. A compliance file for an AI-enabled tool will need to explain how the clinician can evaluate the recommendation without simply trusting the model. If the answer is that the model's reasoning is too complex to expose in a usable way, the 2026 language does not solve the problem.
Time-Critical Use Was Moved, Not Made Easy
FDA removed language that treated time-critical decision-making as automatically outside Criterion 3, a change several legal analyses correctly describe as notable.[2][4] But the practical problem reappears under Criterion 4: can the healthcare professional independently review the basis for the recommendation in the time available?[1]
That relocation is not cosmetic, but it is also not a free pass. A tool used during a slow medication reconciliation review and a tool used during a rapidly deteriorating inpatient event may present the same kind of recommendation on paper. The second tool gives the clinician far less time to inspect the basis, compare alternatives, and decide whether the software is wrong.
For AI CDS teams, the regulatory question is therefore less about whether a workflow is labeled urgent and more about whether independent review is realistic in that workflow. A dashboard that allows a clinician to inspect the relevant labs, vitals, source guideline, and reasoning path before acting is in a different position from an alert that fires in a crisis with a model score and little else. The guidance gives developers a better place to make the argument, not a guarantee that FDA will accept it.
Criterion 4 Is Softer, But It Still Carries the AI Burden
Criterion 4 has always been the pressure point for CDS that wants to avoid device treatment: the healthcare professional must be able to independently review the basis for the recommendation rather than rely primarily on the software. In the 2026 revision, FDA softened the transparency expectations by emphasizing information that is usable for the intended user rather than exhaustive disclosure of every technical detail.[1][2][4]
That is a sensible correction. A 40-page model appendix is not independent review if the clinician cannot use it during care. The relevant question is whether the user receives enough meaningful information to understand why the recommendation appeared and whether it fits the patient in front of them.
For many AI-enabled CDS products, this will push teams toward practical transparency artifacts: the patient-specific inputs used, the clinical source or rule the output depends on, the factors most responsible for a risk estimate, known limitations, and clear instructions about what the software is and is not intended to do. None of those items is magic. Together, they can help show that the clinician is reviewing a basis, not just ratifying a prediction.
The uncomfortable part is that usability and sufficiency are not the same thing. FDA's softer language may reduce the temptation to overload clinicians with technical disclosures, but it does not define how much explanation is enough for non-deterministic models. A model that produces different answers from similar prompts, or that generates prose rather than a stable score, makes the Criterion 4 question much harder than a rules-based drug interaction checker.
Documentation Tools Get a Clearer Opening
The revised guidance also adds examples of clinical documentation tools that may qualify for enforcement discretion, including an AI tool that analyzes a radiologist's clinical findings to generate a proposed radiology report summary.[1][3][2] This is one of the more concrete signals in the document, and it matters because documentation-adjacent AI is already easier to deploy operationally than many diagnostic AI products.
The boundary is still important. A system that drafts a summary from clinician findings is different from a system that identifies a lesion, determines its clinical significance, and tells the radiologist what diagnosis to make. The first may support documentation. The second begins to look like diagnostic decision support, and possibly device functionality, depending on its claims and use.
This is where product language can become a regulatory fact. If marketing says the tool improves diagnostic accuracy, prioritizes urgent cases, or detects disease that the clinician might miss, the company has moved away from a modest documentation theory. If the tool proposes a report summary based on already-entered clinical findings and leaves the clinician responsible for review, the 2026 examples provide more useful footing.
The Guidance Is Silent Where AI Is Noisiest
The most striking omission is generative AI. The revised CDS guidance does not address generative AI, LLM chatbots, or the specific problem of applying independent review to outputs that may be probabilistic, variable, conversational, or prompt-sensitive.[2] That silence is hard to square with public framing that treated the revision as part of the AI policy story.

A chatbot-style CDS tool does not behave like a conventional calculator. It may answer follow-up questions, reframe recommendations, introduce unsupported reasoning, or vary its response across sessions. Even if a clinician is the intended user, the independent-review analysis becomes more demanding: what exactly is the recommendation, what basis is being reviewed, and how does the developer ensure the user can distinguish grounded clinical support from fluent explanation?
The 2026 guidance does not answer those questions. It gives teams familiar CDS criteria to apply, but it does not tell them how FDA will evaluate LLM-specific behavior. That leaves product counsel with a familiar but unsatisfying memo structure: describe the intended use narrowly, map each function to the CDS criteria, document transparency controls, and then flag that FDA has not yet provided AI-specific treatment for the model architecture.
Consumer-facing tools remain another unresolved area. FDA had proposed enforcement discretion for some consumer-facing CDS in a 2022 draft, but that approach was not finalized, and the 2026 revision does not create a new safe harbor for symptom checkers or health chatbots used directly by patients.[2] That matters because the independent-review premise is structurally different when the user is not a licensed healthcare professional.
Hospital-developed predictive CDS sits in a separate gray zone. Academic medical centers and health systems have developed models for use cases such as sepsis, deterioration, and opioid risk, and the regulatory status of these locally developed tools has been a recurring concern in the hospital medicine literature.[7] The January 2026 guidance does not provide a tailored answer for that class of models.
Device AI Is Expanding While CDS Policy Is Being Reworked
The CDS update lands against a busy AI device backdrop. Innolitics' independent year-in-review analysis counted 295 AI/ML-enabled medical device clearances in 2025, a record in its database, with a median 510(k) clearance time of 142 days.[5] The same analysis reported that 30 of those 295 devices, or 10%, included predetermined change control plans for iterative updates.[5]
Those figures are useful context, not proof that the CDS guidance will accelerate adoption. They describe cleared or authorized devices, while the CDS guidance is largely about when certain software functions may avoid device regulation or receive enforcement discretion. Still, they show why the guidance matters now: FDA is revising software policy while AI medical devices are no longer a marginal category.
The same Innolitics analysis found that radiology accounted for 211 of 295 AI/ML clearances in 2025, or 71.5%, followed by cardiovascular at 8.8% and neurology at 4.7%.[5] That concentration helps explain why the new radiology-report-summary example will attract attention, but it should not be overread. A documentation summary example is not a diagnostic imaging clearance pathway.
A broader Bipartisan Policy Center analysis reported more than 1,250 AI-enabled medical devices authorized in the United States as of mid-2025, with 96% through 510(k), 3% through De Novo, and 1% through PMA.[6] The center notes that FDA's public database identifies AI-enabled devices primarily through AI-related terms in summary descriptions, so the count is useful but not necessarily comprehensive.[6]
What Developers Can Responsibly Take From the 2026 Guidance
The practical takeaway is not that AI CDS has been deregulated. It is that certain AI-enabled tools now have a better argument if they are designed around the actual CDS criteria rather than around the hope that being software makes them low risk.
- For a single-output tool, document why only one option is clinically appropriate and how the user can review the basis for that output.
- For a time-sensitive tool, test whether independent review is feasible in the real workflow, not just theoretically possible in a training deck.
- For an AI documentation function, keep claims aligned with drafting, summarization, and clinician review unless the product is prepared for a different regulatory posture.
- For an LLM-enabled CDS product, assume the absence of AI-specific guidance is a risk factor, not an invitation to skip the device analysis.
- For a consumer-facing health chatbot, do not rely on the 2026 CDS guidance as if it created enforcement discretion for patient-directed advice.
This is also the point where governance has to get specific. A health system buying an AI CDS tool should ask for the intended-use statement, the criteria analysis, the evidence supporting the recommendation logic, the clinician-facing explanation, and the vendor's position on whether the function is a device. A developer should expect those questions before procurement, not after an incident.
The Next Phase Is Promised, Not Settled
Commissioner Marty Makary has previewed a deregulatory direction for FDA software policy, including eliminating at least half of existing software and digital health guidances and developing a new risk-based AI framework with emphasis on post-market monitoring.[3] Those statements are important signals. They are not yet binding policy.
FDA also plans to revise its Policy for Device Software Functions and Mobile Medical Applications in fiscal year 2026, which could affect how software functions adjacent to CDS are treated.[2] Separately, FDA has already issued guidance on marketing submission recommendations for predetermined change control plans for AI-enabled device software functions, reflecting the agency's ongoing work on iterative AI systems.[8]
The January 2026 CDS guidance therefore sits in an interim position. It reduces friction for some AI-enabled CDS and documentation-adjacent tools. It gives developers more room around single clinically appropriate recommendations and more realistic transparency expectations. It does not decide how generative AI, conversational CDS, consumer health advice, or hospital-developed predictive models should be regulated.
Until FDA issues the promised risk-based AI framework or revises related software guidance, the safest reading is also the most useful one: treat the 2026 CDS guidance as targeted relief within the existing CDS structure, not as the AI policy reset itself.
References
- Clinical Decision Support Software Final Guidance, FDA, January 2026.
- 5 Key Takeaways from FDA's Revised CDS Software Guidance, Covington.
- FDA Cuts Red Tape on Clinical Decision Support Software, Arnold & Porter.
- A Relaxing 2026? FDA Updates General Wellness and CDS Software Guidance, Jones Day.
- 2025 Year in Review — AI/ML Medical Device 510(k) Clearances, Innolitics.
- FDA Oversight: Understanding the Regulation of Health AI Tools, Bipartisan Policy Center.
- Regulatory Oversight of Clinical Decision Support Systems: A New Era for Health Care AI?, Journal of Hospital Medicine, 2021.
- Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions, FDA, August 2025.
Comments
Join the discussion with an anonymous comment.