AI medical device lifecycle management is no longer a future-state policy topic. By Q3 2026, manufacturers are trying to reconcile three moving pieces at once: FDA’s January 2025 draft recommendations for AI-enabled device software functions, IMDRF’s draft N93 technical framework with consultation closing on July 10, 2026, and the EU AI Act’s core high-risk AI obligations becoming fully enforceable in August 2026.[1][2][3] Two of those instruments are still draft or consultative. One is a horizontal AI law that sits beside, not inside, the medical device regulation. The practical problem is that the manufacturer still has one quality system, one engineering organization, one clinical or performance evaluation function, and one post-market monitoring operation.
That is why the convergence matters. Not because the frameworks are interchangeable; they are not. FDA remains anchored in device marketing submissions and device software functions. IMDRF N93 is a draft harmonization framework. The EU AI Act adds high-risk AI obligations alongside MDR conformity responsibilities. But across those differences, the regulatory center of gravity is moving toward the same operational expectation: AI risk is managed through the full lifecycle, with evidence that survives design review, submission review, Notified Body scrutiny, and post-market performance drift.
The scale is no longer theoretical. FDA officials stated that the agency had authorized more than 1,000 AI-enabled devices through established premarket pathways as of January 2025, with cumulative authorizations reaching approximately 1,451 by the end of 2025.[4] For teams tracking the authorization landscape, the existing FDA AI-enabled medical devices database is useful background. The harder work now is not counting cleared products. It is deciding which lifecycle controls can be made common, which records need jurisdiction-specific evidence, and where a single SOP will create ambiguity rather than efficiency.
The Shared Core Is Lifecycle Control, Not AI Philosophy
The three frameworks approach AI-enabled devices from different legal and institutional directions, but they put pressure on the same parts of the manufacturer’s operating system. A useful convergence map starts with the controls that quality, regulatory, software, clinical, and post-market teams must actually maintain.
| Lifecycle obligation | Common operational meaning | Where the mapping needs care |
|---|---|---|
| Lifecycle risk management | AI-specific hazards, foreseeable misuse, performance degradation, and residual risks are controlled from design through post-market review. | FDA recommendations are tied to marketing submission content; IMDRF describes lifecycle concepts; EU obligations apply through high-risk AI requirements alongside MDR processes. |
| Data governance and bias mitigation | Training, tuning, validation, and real-world data need documented provenance, suitability, representativeness, and limitations. | Terminology and evidence depth differ; a general data SOP will not usually be enough for all submissions or conformity reviews. |
| Verification, validation, and performance evaluation | Model performance is established before deployment and re-evaluated when data, use conditions, or model behavior change. | FDA submission expectations, IMDRF lifecycle phases, and EU conformity evidence do not collapse into one identical test report. |
| Post-market monitoring and drift detection | Manufacturers define what to monitor, how signals are escalated, and when drift becomes a quality event or change-control trigger. | Monitoring is the area where vague lifecycle language most quickly becomes an audit finding if responsibilities and thresholds are not assigned. |
| Transparency, explainability, and human oversight | Users, reviewers, and oversight personnel receive documentation appropriate to intended use, known limitations, and human decision points. | The concepts overlap, but labels and disclosure expectations vary across frameworks. |
| Cybersecurity and change control | Updates, retraining, environment changes, and threat responses are planned, justified, tested, documented, and linked to risk controls. | FDA’s PCCP mechanism gives one concrete model; EU and IMDRF mapping still requires separate evidence alignment. |
This map is deliberately framed around obligations rather than regulators. A quality manager does not implement “FDA” on Monday, “IMDRF” on Tuesday, and “EU AI Act” on Wednesday. The same model version is trained. The same data pipeline is controlled. The same complaint trend is reviewed. The same performance drift either does or does not trigger escalation. The documentation can be modular, but the underlying control should not be fictional.
IMDRF’s Seven Phases Are a Useful Spine, If They Are Not Treated as Seven Equal Binders
IMDRF draft N93 describes seven AI lifecycle phases: Planning and Design, Data Collection and Management, Model Building and Tuning, Verification and Validation, Model Deployment, Operations and Monitoring, and Real-world Performance Evaluation.[2] That sequence is helpful because it keeps the discussion from stopping at clearance or certification. It is less helpful if a manufacturer turns it into seven parallel documentation packages with repeated language and unclear ownership.
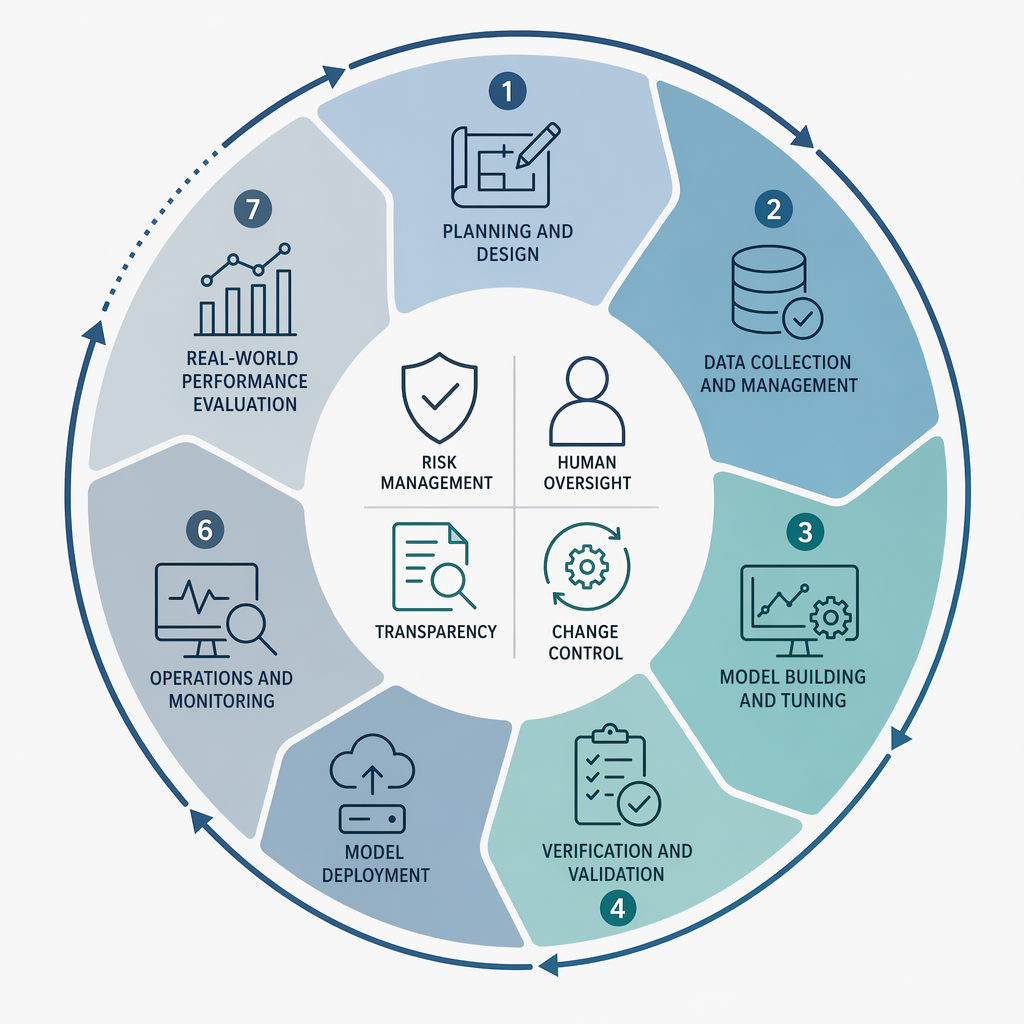
The better use of the seven phases is as a traceability spine. Planning and Design should define the intended use, clinical context, model role, user interaction, foreseeable misuse, and lifecycle change strategy. Data Collection and Management should connect dataset requirements to risk controls and bias mitigations. Model Building and Tuning should preserve traceability between design inputs, model architecture choices, feature handling, and known limitations. Verification and Validation should show that the model meets its predefined performance requirements for the intended population and use conditions. Deployment should control release configuration, installation, user-facing information, cybersecurity assumptions, and monitoring readiness. Operations and Monitoring should make performance signals actionable. Real-world Performance Evaluation should feed the management review, risk file, change-control process, and, when needed, regulatory communication.
The pressure points are not evenly distributed. Planning, data management, validation, monitoring, and change control carry more regulatory weight than a neat lifecycle diagram suggests. If those controls are weak, later claims about transparency, oversight, and safe learning behavior tend to become narrative rather than evidence.
AI Risk Management Has to Be Built Into the Existing QMS
A unified QMS strategy does not mean creating a standalone AI governance committee that reviews principles while the design history file, software lifecycle file, complaint process, and CAPA process continue unchanged. The shared regulatory expectation is narrower and more demanding: AI-specific risk controls must be embedded into the processes that already create auditable evidence.
At design input, the QMS should force the team to state what the model is allowed to do, what the user remains responsible for, what patient groups or acquisition conditions are in scope, and what would make performance unacceptable. At data planning, the same risk file should drive requirements for source data, labeling, curation, representativeness, missingness, and bias evaluation. At validation, the acceptance criteria should not float free of the intended use; they should trace back to clinical or operational claims, user workflow, and risk controls. At deployment, release records should confirm that the model version, software environment, user-facing information, cybersecurity controls, and monitoring configuration match what was validated.
FDA’s draft guidance explicitly covers design, data management, model validation, performance monitoring, cybersecurity, and transparency for AI-enabled device software functions.[1] IMDRF’s draft framework carries the same lifecycle logic through its universal concepts, including QMS integration, risk management throughout the lifecycle, human oversight, cybersecurity, transparency and explainability, and change control.[2] The EU AI Act’s high-risk obligations similarly include risk management, data governance, transparency, human oversight, and post-market monitoring for covered high-risk AI systems, including AI-enabled medical devices that fall within its scope.[3]
The useful question is not whether the QMS has an “AI policy.” It is whether a reviewer can follow one risk control from design planning into dataset specifications, model development records, validation evidence, labeling or user information, release approval, monitoring thresholds, and change-control decisions. If that chain breaks, the manufacturer may still have documents, but it does not yet have lifecycle management.
Data Governance Is Where Bias Controls Become Verifiable
Bias mitigation is often described as if it lives in model evaluation, but most of the auditable work starts earlier. The QMS should require dataset purpose, inclusion and exclusion logic, source controls, annotation procedures, data quality checks, subgroup analysis planning, and documented limitations. A validation report that discovers a gap late may be honest, but it does not substitute for design-stage data requirements.
For FDA-facing teams, this connects directly to the agency’s draft lifecycle recommendations and the separate practical discussion of transparency and bias under FDA’s 2025 AI device draft guidance. For EU-facing teams, it also has to be readable in the language of high-risk AI data governance. For IMDRF alignment, it should map back to Data Collection and Management, Model Building and Tuning, Verification and Validation, and Real-world Performance Evaluation rather than appearing in only one lifecycle phase.
Monitoring Is the Operational Core, Not a Postscript
The lifecycle frameworks become most concrete after deployment. That is where a model encounters new scanners, software dependencies, site practices, population shifts, adversarial pressure, workflow shortcuts, and user behavior that did not appear cleanly in premarket testing. A monitoring plan that only says “track performance” is not a control. It is an intention.
A defensible monitoring architecture should identify the monitored signals, data sources, frequency of review, statistical or operational thresholds, escalation pathway, responsible function, and link to risk management. Some signals may be technical, such as input distribution changes or failed processing. Some may be clinical or performance-related, such as changes in sensitivity, specificity, alert burden, or user override patterns, depending on the device and its claims. Some may come through complaint handling, service records, cybersecurity monitoring, or field performance evaluation. The point is not to collect everything; it is to define which evidence would show that the device is no longer performing as expected.
This is also where drift detection has to be more than a data science dashboard. If drift is detected, someone must decide whether it is expected variation, a degraded performance signal, a usability issue, a cybersecurity issue, a data pipeline issue, or a trigger for retraining or other modification. That decision should not depend on the availability of the one engineer who understands the model. It belongs in documented procedures with review authority, records, and escalation criteria.
The FDA, Health Canada, and MHRA Good Machine Learning Practice principles include monitoring deployed models and managing re-training as a lifecycle concern, with Principle 10 focused on monitoring performance and managing re-training risks.[7] That principle sits comfortably behind FDA’s TPLC draft guidance, IMDRF’s Operations and Monitoring and Real-world Performance Evaluation phases, and the EU AI Act’s post-market monitoring expectations. It does not, by itself, tell a manufacturer what threshold to set. That remains device-specific and risk-based.
The failure mode is familiar: premarket evidence is treated as the real regulatory file, while post-market monitoring is handled as a periodic report assembled from whatever data happened to be available. For AI-enabled devices, that split is becoming harder to defend. The practical challenges are explored further in why post-market surveillance fails for AI medical devices, but the regulatory direction is already clear enough: monitoring has to be designed before deployment, not improvised after the first unexpected trend.
PCCP Shows What Controlled Learning Can Look Like
FDA’s Predetermined Change Control Plan mechanism deserves more than a footnote because it converts lifecycle management into a submission artifact. It is one of the clearest examples of how a regulator can allow certain future modifications without treating every anticipated update as an uncontrolled surprise.
The FDA, Health Canada, and MHRA issued guiding principles for PCCPs for machine learning-enabled medical devices, and FDA finalized guidance in 2025 describing PCCP marketing submission recommendations for AI-enabled device software functions.[5][6] The FDA final guidance identifies three required components: a Description of Modifications, a Modification Protocol, and an Impact Assessment.[6] Approximately 53 AI/ML-enabled devices had been authorized with PCCPs as of the end of 2024.[6]
| PCCP component | What it forces the manufacturer to specify | Why it matters for lifecycle convergence |
|---|---|---|
| Description of Modifications | The planned future changes, within defined boundaries. | Prevents “continuous learning” from becoming an undefined permission to change the device. |
| Modification Protocol | The methods, data, testing, acceptance criteria, and implementation controls for the planned changes. | Connects retraining or updating to validation, risk management, cybersecurity, release control, and documentation. |
| Impact Assessment | The expected effect of the modifications on safety and effectiveness. | Creates a structured link between change planning and the evidence needed to justify continued control. |
PCCP is not a universal shortcut, and it is not an EU AI Act compliance plan. Its value is more specific: it demonstrates the kind of predefined boundary-setting that lifecycle governance needs. If the model may be updated, the manufacturer should be able to say what kinds of updates are allowed, what data may be used, what testing will be performed, what acceptance criteria apply, who approves release, how users are informed when necessary, and what post-update monitoring will confirm.
That structure travels well even where the legal mechanism does not. A unified QMS can use the PCCP logic as a change-control pattern: define permissible change categories, link each category to validation and risk controls, require documented impact assessment, and preserve jurisdiction-specific submission or notification decisions. The common control is planned change governance. The regulatory output may still differ by market.
Transparency and Human Oversight Need Evidence, Not Just User-Friendly Language
Transparency, explainability, and human oversight appear across the frameworks, but the terms should not be treated as interchangeable labels pasted onto the same paragraph. FDA’s draft guidance includes transparency among its lifecycle topics.[1] IMDRF draft N93 identifies transparency and explainability, as well as human oversight, among its universal concepts.[2] The EU AI Act’s high-risk obligations include transparency and human oversight requirements.[3]
For a manufacturer, the evidence question is practical. What must the reviewer know to evaluate the device? What must the user know to use it safely? What must the oversight function know to detect misuse, automation bias, or performance degradation? What information must be preserved internally to investigate a complaint, deviation, or field signal? Those are not always the same audience, and a single public-facing explanation rarely satisfies all of them.
A traceable approach separates at least three records: reviewer-facing documentation that explains model development, validation, limitations, and risk controls; user-facing information that supports safe use and appropriate reliance; and internal QMS records that support investigation, monitoring, change control, and management review. Teams comparing terminology across markets may also benefit from a more focused comparison of explainable AI medical device requirements across regulators.
Where the Frameworks Do Not Collapse Into One Another
The case for a unified QMS is strong enough to act on, but not strong enough to erase regulatory boundaries. Treating the three frameworks as identical creates its own compliance risk.
- FDA’s January 2025 lifecycle guidance remains draft as of July 2026, so it should inform preparation but should not be described internally as final FDA policy.[1]
- IMDRF N93 is a draft technical framework published April 7, 2026, with public consultation closing July 10, 2026; it may change before finalization.[2]
- The EU AI Act creates high-risk AI obligations that apply alongside existing MDR conformity responsibilities, producing dual conformity assessment considerations rather than a simple replacement of device regulation.[3]
- The EU AI Act has phased timing, including the August 2026 enforceability of core high-risk AI obligations and later timing for some CE-marked devices already under Notified Body review.[3]
- Terminology differs across the instruments, especially around transparency, explainability, lifecycle monitoring, and change control; conceptual mapping does not mean identical evidence expectations.
Those distinctions affect real work. A design control record may support all three frameworks, but an FDA marketing submission section, an IMDRF-aligned lifecycle matrix, and an EU technical documentation package may need different wording, cross-references, and emphasis. A post-market monitoring SOP may be common, but reporting triggers and conformity assessment interactions may differ. A change-control board can use one decision tree, but that tree must still branch when market-specific submission, notification, or Notified Body engagement is required.
This is where “alignment” often becomes dangerous. If it means one traceability architecture with controlled jurisdictional outputs, it can reduce duplicated effort. If it means one generic checklist labeled FDA, IMDRF, and EU, it will fail at the first serious review question.
What a Unified Traceability System Should Actually Contain
The most durable architecture is a requirements and evidence matrix that sits above individual submissions but is specific enough to drive them. It should not merely list regulatory clauses. It should connect lifecycle obligations to QMS procedures, product records, responsible owners, review gates, and market-specific evidence.
| Traceability layer | Purpose | Example evidence |
|---|---|---|
| Lifecycle obligation | Defines the shared control area across frameworks. | Risk management, data governance, validation, monitoring, transparency, human oversight, cybersecurity, change control. |
| QMS procedure | Shows where the obligation is operationalized. | Design control SOP, software lifecycle SOP, data management SOP, post-market surveillance SOP, CAPA procedure, change-control procedure. |
| Product record | Shows device-specific implementation. | Risk file, dataset specification, model development record, validation protocol and report, release record, monitoring plan. |
| Review trigger | Shows when reassessment is required. | Performance drift, dataset change, retraining proposal, cybersecurity event, complaint trend, intended-use change. |
| Market-specific output | Shows how common evidence is translated for each framework. | FDA submission content, IMDRF lifecycle mapping, EU technical documentation and conformity assessment evidence. |
The matrix should be maintained under document control, not treated as a regulatory affairs spreadsheet that only appears before a submission deadline. When a model is retrained, a dataset is expanded, a monitoring threshold is revised, or a new market is added, the matrix should show which records are affected and which market-specific obligations must be reconsidered.
This also helps resolve ownership. Data science owns some records, but not the QMS. Regulatory owns submissions, but not every monitoring signal. Quality owns procedures and records, but cannot define model acceptance criteria alone. Clinical, cybersecurity, human factors, software engineering, and post-market teams all touch the evidence chain. A unified architecture makes those dependencies visible before an auditor or reviewer does.
The Compliance Strategy for 2026 Is Bounded Convergence
For AI medical device lifecycle management, convergence is real enough to justify building one QMS architecture. The shared controls are now too consistent to maintain three unrelated compliance tracks without wasting effort and increasing the chance of contradictions. Risk management, data governance, validation, monitoring, drift detection, transparency, human oversight, cybersecurity, and change control should be designed as common lifecycle controls.
The convergence is also bounded. FDA’s draft TPLC recommendations, IMDRF’s draft lifecycle framework, and the EU AI Act’s high-risk obligations differ in legal status, terminology, timing, and conformity assessment consequences. A manufacturer that ignores those differences is not harmonizing; it is hiding variance until review.
The defensible path is a single traceability system that maps shared lifecycle controls to each framework’s language and evidence expectations. That system should let a quality manager answer four questions without opening three unrelated binders: what control applies, where it is implemented in the QMS, which product evidence proves it, and what changes when the device enters a different jurisdiction or lifecycle state.
References
- Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations, FDA, January 2025.
- Technical Framework for Artificial Intelligence Life Cycle Management, IMDRF, April 7, 2026.
- EU AI Act 2026: What AI-Enabled Medical Device Manufacturers Need to Know Now, Daiki.
- A Lifecycle Management Approach toward Delivering Safe, Effective AI-enabled Health Care, FDA.
- Predetermined Change Control Plans for Machine Learning-Enabled Medical Devices: Guiding Principles, FDA/Health Canada/MHRA.
- Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions, FDA, 2025.
- Good Machine Learning Practice for Medical Device Development: Guiding Principles, FDA/Health Canada/MHRA.
Comments
Join the discussion with an anonymous comment.