Only 26% of EU hospital representatives surveyed in a European Commission study said they felt ready for AI Act obligations, and the first high-risk classification provisions begin to matter operationally from August 2026.[1][2] For healthcare facilities, the practical question is no longer whether the EU AI Act is relevant. It is: which AI systems in clinical or administrative workflows are high-risk, who owns the deployer controls, and what evidence will the organization be able to produce when someone asks two years after go-live?
This article focuses on healthcare facilities acting as deployers: hospitals, clinics, diagnostic centers, laboratories, and EU-facing care organizations using AI systems in practice. It does not try to restate the entire EU AI Act or cover manufacturer compliance in full. The narrower issue is harder to delegate: when a facility uses a high-risk AI system, it must be able to show that human oversight, logging, training, risk assessment, transparency, monitoring, and incident processes are not just vendor slideware but working controls inside the organization.

The Compliance Clock Is Already Running
The enforcement picture is phased, which is useful for planning and dangerous if treated as permission to wait. Three dates matter for healthcare deployers: Title I-III provisions, including classification rules, become effective in August 2026; deployer obligations for high-risk systems that are also MDR or IVDR Class III devices apply from August 2027; and full deployer obligations for all high-risk systems apply from August 2028, subject to possible changes from the Digital Omnibus and MDR revision proposals.[2]
| Timing | What healthcare facilities should treat as the operational trigger |
|---|---|
| August 2026 | Classification rules and related Title I-III provisions become effective; facilities need an inventory and classification rationale, not just a list of AI pilots. |
| August 2027 | Deployer obligations begin for high-risk systems that are also MDR or IVDR Class III devices. |
| August 2028 | Full deployer obligations apply to all high-risk systems, unless final legislative changes alter the timeline. |
The dates do not map neatly onto hospital project plans. A radiology procurement, an EHR-integrated prediction model, an ambient documentation tool, and a laboratory triage system may each move through contracting, validation, cybersecurity review, DPIA work, clinician training, and go-live at different speeds. If classification waits until legal review at the end of procurement, the facility has already lost the chance to shape the contract around logs, oversight instructions, data retention, incident cooperation, and access to technical documentation.
Start With Scope, Not With the Vendor’s Label
A healthcare facility is usually a deployer when it uses an AI system under its authority in a clinical, diagnostic, laboratory, administrative, or patient-facing workflow. That includes systems bought from vendors, integrated through cloud platforms, embedded in imaging equipment, added to an EHR module, or introduced as a productivity tool by a department. The deployer label matters because Article 26 places obligations on users of high-risk AI systems, including using the system according to instructions, assigning human oversight, keeping logs where under the deployer’s control, and monitoring operation.[3]
Classification is the first place where a clean governance program can become messy. Under the AI Act, high-risk status can arise through the Article 6(1) route, where the AI system is a safety component of a product or is itself a product covered by sectoral legislation such as the MDR or IVDR, and through the Article 6(2) route, where the system falls within Annex III use cases.[3] In healthcare, those routes can overlap with familiar categories, but they do not always behave like the categories a hospital already uses for medical devices, software, clinical decision support, or documentation tools.
Clinical decision support is the obvious difficult case, but it is not the only one. Ambient AI scribes, coding support, referral prioritization, patient risk stratification, scheduling tools, quality-measure abstraction, and LLM-based summarization may have very different risk profiles depending on what they influence and how staff rely on them. A tool that only formats a note is not the same as a tool that changes diagnostic suspicion, treatment priority, or access to care. The facility needs a classification record that explains the actual workflow, not a generic statement that the product is “administrative” or “not diagnostic.”
The European Commission’s high-risk classification guidance was still evolving in 2026, with RAPS reporting on draft guidelines and related uncertainty around classification and possible legislative adjustment.[4] That uncertainty should change the tone of the record, not remove the need for one. A defensible classification file can say what was known at the time, which route was assessed, which use case was assumed, what vendor information was relied on, and what event would trigger reclassification.
A 10-Step Roadmap Facilities Can Actually Assign
A June 2026 Frontiers in Digital Health article by Dennstädt and colleagues proposes a 10-step compliance framework for healthcare facilities. It is not binding Commission guidance, but it is a useful expert synthesis because it translates deployer obligations into work packages that can be assigned to governance, DPO, IT security, quality, clinical leadership, and procurement teams.[1]
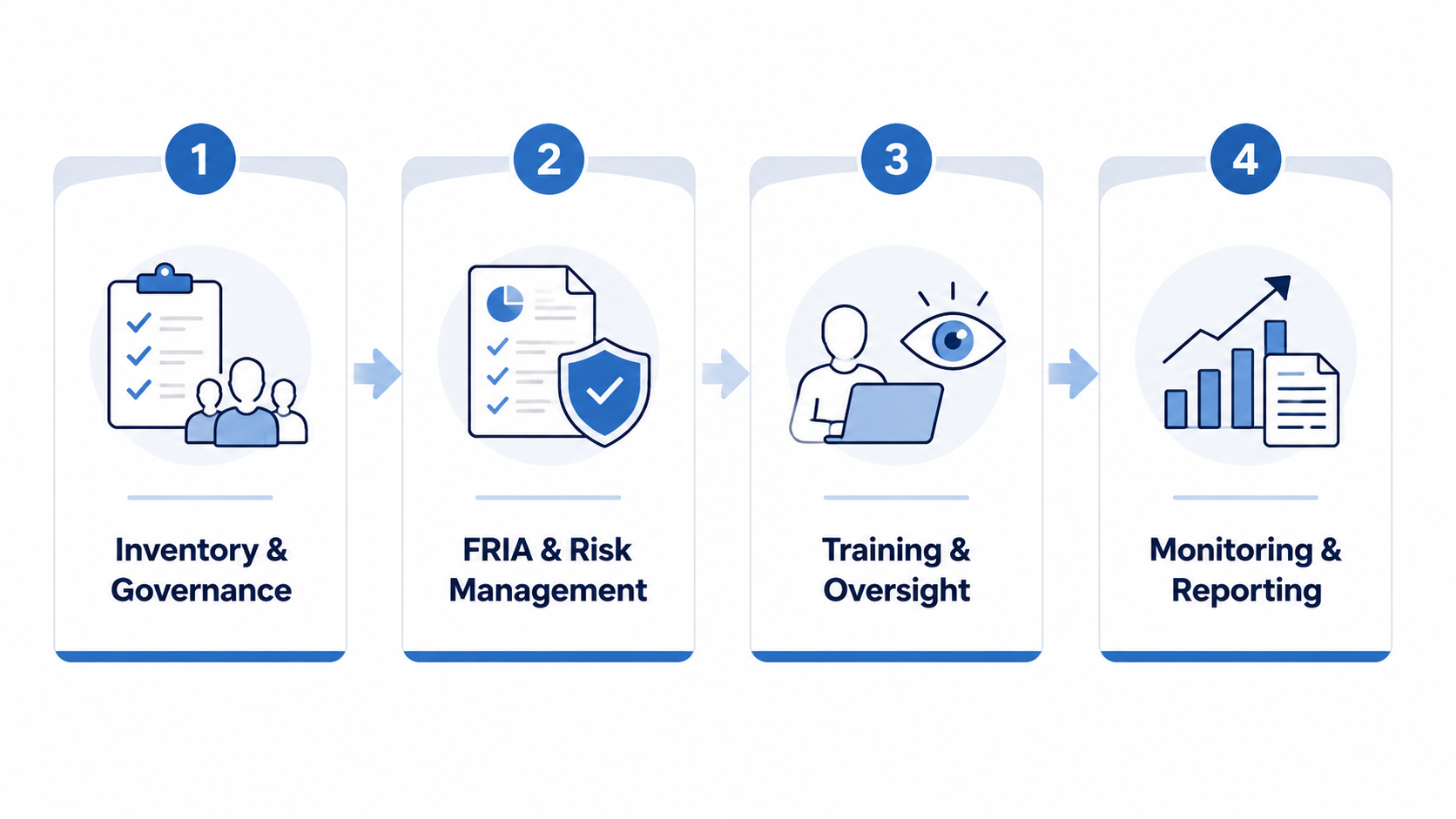
| Phase | Core work |
|---|---|
| Phase 1 | Create an AI governance committee; build an inventory; classify systems. |
| Phase 2 | Perform Fundamental Rights Impact Assessments; connect AI risk management with GDPR, clinical risk, cybersecurity, and technical-documentation review. |
| Phase 3 | Deliver AI literacy training; implement human oversight mechanisms; handle transparency and communication. |
| Phase 4 | Manage registration and reporting; maintain logs, incident workflows, and continuous monitoring. |
The table makes the roadmap look orderly. In practice, the first two phases carry most of the hidden labor. A facility that does not know where AI is already being used cannot classify it. A facility that cannot explain lawful basis, special-category data processing, retention, controllership, and cloud transfers cannot complete a healthcare-grade rights assessment by stapling an AI checklist to an old DPIA.
Phase 1: Governance, Inventory, and Classification
The governance committee does not need to be a new empire. It does need enough authority to stop AI decisions from scattering across procurement, research, radiology, pathology, innovation, and digital transformation with no shared register. A practical committee usually needs clinical leadership, quality and patient safety, DPO or privacy, IT security, legal, procurement, medical device or software governance, and the service line that owns the workflow.
The inventory should begin before procurement finalization and should include systems already embedded in clinical workflows. The uncomfortable items are often not the flagship AI products. They are add-on modules in imaging platforms, risk scores activated inside enterprise software, documentation assistants trialed by a department, lab workflow tools, or vendor updates that introduce AI functionality after the original contract was approved.
- System name, vendor, version, deployment location, and clinical or administrative workflow.
- Purpose, intended users, affected patient or staff groups, and whether output informs diagnosis, treatment, triage, prioritization, eligibility, documentation, coding, or resource allocation.
- Classification rationale under Article 6(1), Article 6(2), Annex III, or a documented non-high-risk conclusion.
- Provider information, CE or conformity status where relevant, technical documentation received, instructions for use, and known limitations.
- Data categories processed, integration points, cloud or third-party dependencies, logging availability, and local owner.
- Review date, trigger events for reassessment, and the committee decision record.
Classification should not be left as a single drop-down field. If the tool is treated as not high-risk, the file should explain why. If the conclusion depends on a narrow use, the facility should document that boundary and train against drift. A summarization tool used only to draft a visit note may be assessed differently from the same tool used to generate differential diagnoses or referral priority. The record must follow actual use, not the safest sentence in the product brochure.
Phase 2: FRIA, GDPR, Risk Management, and Technical Evidence
For deployers of certain high-risk systems, Article 27 requires a Fundamental Rights Impact Assessment before use.[3] In healthcare, that assessment cannot be treated as a generic fairness note. It has to sit beside patient safety, data protection, equality, access to care, clinical accountability, and operational resilience. The Frontiers framework specifically connects the FRIA to GDPR lawful basis under Articles 6 and 9, retention periods, joint-controllership analysis, and cross-border transfer audits for cloud-based AI systems.[1]
That connection matters because health data is special-category data, and AI projects often change more than one processing condition at once. A vendor may host model inference in the cloud, store prompts or outputs for service improvement, rely on subprocessors outside the EU, or require access to pseudonymized training or monitoring data. The DPO cannot answer the FRIA question without seeing the data-flow diagram, retention schedule, security controls, role allocation, and contract terms.
| FRIA question | Healthcare evidence that should exist |
|---|---|
| Who may be affected? | Patient groups, staff groups, and any subgroup that could experience reduced access, delayed care, inappropriate prioritization, or disproportionate surveillance. |
| What decision or workflow is influenced? | A workflow map showing where the AI output appears, who sees it, and whether it can change diagnosis, treatment, triage, documentation, coding, scheduling, or resource allocation. |
| What GDPR basis applies? | Documented lawful basis under Article 6 GDPR, special-category condition under Article 9 GDPR, and any local legal basis relied on for healthcare processing. |
| Who controls the processing? | Controller, processor, or joint-controller analysis, including vendor roles and any secondary use of data. |
| Where does data go? | Cloud architecture, subprocessors, cross-border transfer assessment, retention periods, deletion duties, and audit rights. |
| How is harm detected? | Monitoring metrics, complaint channels, clinical escalation routes, incident thresholds, and review frequency. |
Technical documentation review is another place where deployer work depends on provider cooperation. A hospital may not have to create the manufacturer’s full technical file when it is only deploying a vendor system, but it still needs enough information to use the system safely and prove that its own obligations were realistic. Instructions for use, intended purpose, limitations, performance information, oversight requirements, logging capabilities, cybersecurity dependencies, and update procedures should be reviewed before signature, not requested after the first adverse event.
Risk management should not duplicate every existing committee. It should connect them. If the AI system is also a medical device, medical device governance and clinical safety review will already carry part of the burden. If it uses patient data at scale, DPIA and cybersecurity review will carry another part. If it changes staff workflow, quality and training evidence become relevant. The AI governance record should show how these streams meet, what each owner decided, and what residual risk the facility accepted.
Phase 3: AI Literacy, Human Oversight, and Communication
Article 4 requires providers and deployers to take measures to ensure a sufficient level of AI literacy among staff and others dealing with AI systems on their behalf.[3] For a hospital, attendance at one general AI webinar will not be convincing evidence. Training should match the system and the role: a radiologist reviewing CAD output needs different instruction from a ward clerk using a summarization tool, a laboratory supervisor monitoring queue prioritization, or an IT analyst reviewing logs.
- Clinical users should understand intended purpose, known limitations, override expectations, escalation routes, and when not to rely on the output.
- Operational users should understand workflow boundaries, documentation duties, patient communication scripts where applicable, and how to report unusual behavior.
- IT and security teams should understand integration points, access control, logging, update management, and vendor incident contacts.
- Governance and quality teams should understand monitoring indicators, review cadence, complaint handling, and reclassification triggers.
Human oversight under Article 26(2) needs a named mechanism, not a ceremonial statement that “the clinician remains responsible.”[3] The facility should identify who reviews outputs, when review is mandatory, what the user can override, how disagreement is documented, and who has authority to suspend use. Oversight also has to be plausible within staffing reality. A policy that requires detailed human review but gives staff no time, interface cues, or escalation path is not an operational control.
Transparency and communication obligations depend on context. Some tools require staff-facing disclosure and instructions; others may affect patient communication, consent language, complaint handling, or access requests. The facility should decide which patient-facing materials, clinician scripts, and internal notices are needed before deployment. This is where a compliance file can look complete while the front desk, ward team, or outpatient clinic is still improvising.
Phase 4: Logs, Reporting, Incidents, and Monitoring
Article 26(6) requires deployers to keep logs automatically generated by high-risk AI systems to the extent such logs are under their control.[3] That phrase needs attention in contracting and architecture. If the vendor controls all logs, the facility still needs access terms, retention expectations, export formats, and incident-support commitments. If the hospital controls local logs, IT must know what is captured, how long it is retained, who can access it, and how it will be protected as clinical or operational evidence.
Registration and reporting duties should be mapped system by system. Some obligations sit with providers; some fall on deployers; some depend on whether the system is high-risk and how it is placed on the market or put into service. The useful hospital artifact is a reporting matrix: who reports what, to whom, within which internal deadline, using which evidence, and with which vendor contact. Waiting until an incident occurs to discover whether quality, legal, IT security, the DPO, or the clinical service owns the report is poor control design.
Continuous monitoring should include more than uptime. A high-risk AI system can remain technically available while drifting clinically, being used outside its intended purpose, producing unexplained output patterns, creating staff workarounds, or affecting one patient group differently from another. Monitoring indicators should be chosen during implementation, reviewed by the governance committee, and tied to suspension or escalation criteria.
Penalties Are Not the Main Work, but They Explain the Stakes
The AI Act’s penalty scale is large enough to be noticed: up to EUR 15 million or 3% of global annual turnover for certain high-risk non-compliance, and up to EUR 35 million or 7% for prohibited AI practices.[3] Those numbers should not become the whole conversation. The more immediate risk for a healthcare facility is a quieter one: a system goes live, the pilot team moves on, a vendor update changes behavior, logs are inaccessible, the FRIA was never completed, and no one can show who trained the staff who now rely on the output.
Most of the controls that reduce that risk are ordinary governance artifacts: an inventory, classification record, FRIA, DPIA linkage, oversight instruction, training roster, logging specification, incident workflow, monitoring dashboard, and committee minutes. None is glamorous. All are easier to build before go-live than reconstruct under pressure.
The In-House Development Trap
A hospital that buys and uses a vendor AI system is usually thinking like a deployer. A hospital that builds an AI system and puts it into service under its own name may have crossed into provider territory. The Johner Institute notes that the AI Act does not recognize the MDR and IVDR concept of in-house manufacturing in the same way; a hospital developing and placing an AI system into service under its own name can become a provider with obligations including CE marking, technical documentation, and notified body conformity assessment.[5]
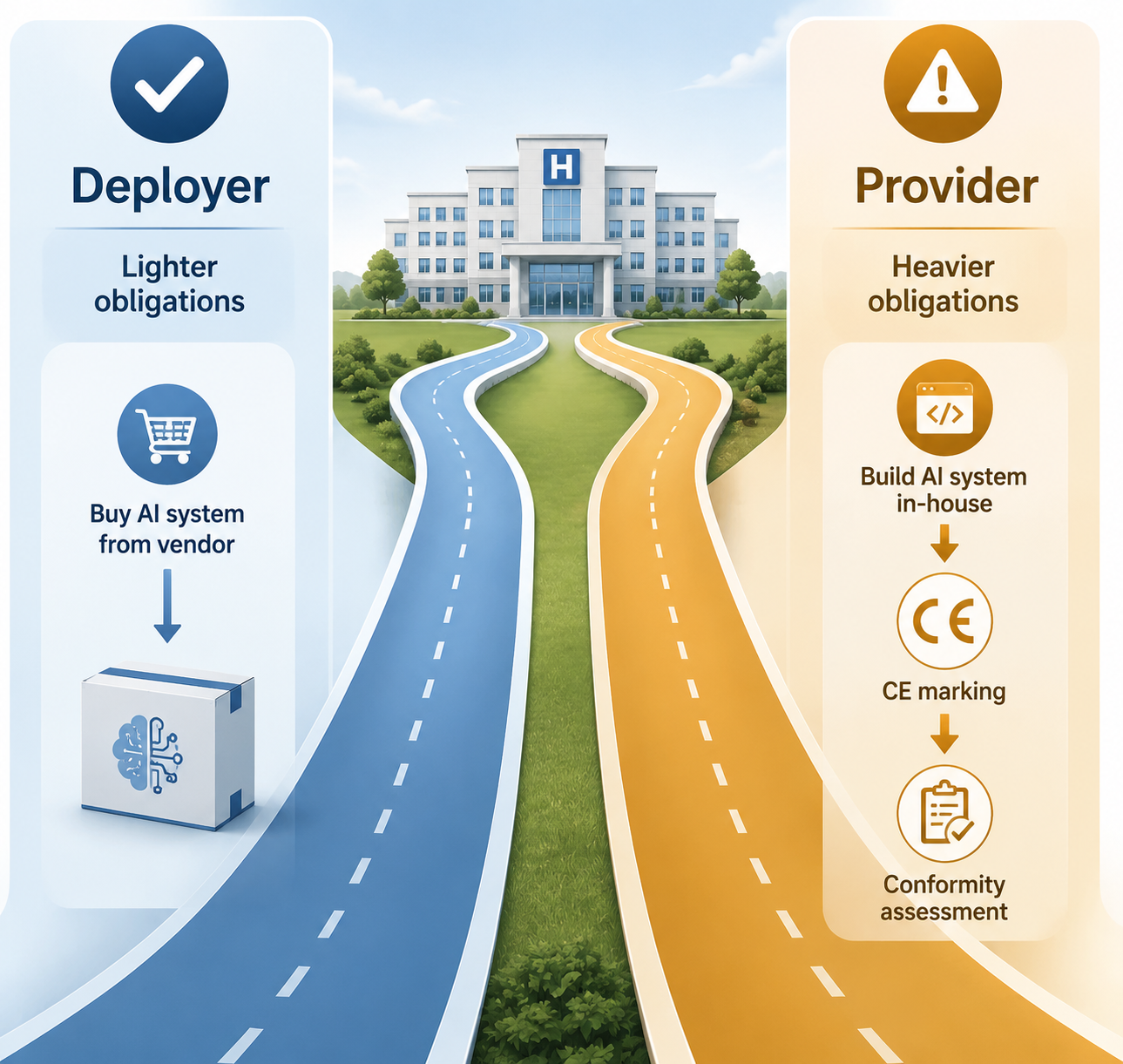
This is the assumption that should make internal innovation programs pause. A local model trained by a data science team, a prediction tool built for one specialty, or an LLM workflow assembled from cloud components may feel administratively safer than procurement because there is no vendor sales cycle. Under the AI Act, that comfort can be false. The organization may be taking on provider obligations it is not staffed to meet.
The practical response is not to ban internal development. It is to route it through the same intake process before clinical use: intended purpose, classification, medical device status, provider versus deployer role, technical documentation plan, quality management expectations, cybersecurity review, data protection review, and conformity assessment dependency. Research prototypes, quality-improvement analytics, and clinical tools put into routine service are not the same governance event.
What Remains Unsettled in July 2026
Several important mechanics are still unsettled. As of July 2026, no notified bodies had been designated under the AI Act, while Germany’s KI-MIG identifies the Federal Network Agency as the central market surveillance body.[1][5] That does not make compliance optional. It does mean facilities should document assumptions, external dependencies, and any vendor or conformity-assessment milestones that could affect deployment.
The Digital Omnibus and MDR revision proposals were also not final as of July 2026. Reed Smith’s analysis notes possible changes that could affect timing and reduce duplicative requirements for medical devices, but the proposals should not be treated as adopted law.[2] Draft classification guidance may clarify some hard cases, yet final interpretation can still shift, particularly for clinical decision support, ambient documentation, and LLM-based systems.[4]
The controllable work is already visible. Healthcare facilities can build inventories, assign governance owners, record classification reasoning, prepare FRIAs that actually connect to GDPR and cloud architecture, negotiate access to logs and technical information, train staff by role, and define monitoring and incident workflows. Waiting for every procedural detail to settle will not make those tasks smaller; it will only move them closer to enforcement.
References
- The EU AI Act: implications and compliance guidance for healthcare facilities, Frontiers in Digital Health, June 2026.
- The EU AI Act and medical devices: Navigating high-risk compliance, Reed Smith.
- EU Artificial Intelligence Act, artificialintelligenceact.eu.
- EU Commission drafts guidelines on classifying high-risk systems under the AI Act, RAPS.
- AI Act: EU AI Regulation, Johner Institute.
Comments
Join the discussion with an anonymous comment.