Yes. In the 2025 source set used here, the FDA authorized more AI/ML-enabled medical devices than in any prior year: 295 devices, up from 253 in 2024 and 221 in 2023.[1] The cumulative count reached 1,451 AI/ML medical device authorizations through December 2025 when counted at the authorization level across FDA sources.[2]
That answer needs the qualifier. Innolitics’ 2025 analysis focuses on AI/ML medical device 510(k) clearances, while the IntuitionLabs tracker aggregates cumulative FDA AI medical device authorizations from multiple FDA sources.[1][2] Those are not interchangeable denominators. For a hospital committee, an FDA AI device count does not name a single level of clinical proof, a single review pathway, or a single kind of product.

The 2025 Dashboard
| Measure | 2025 Finding | Why It Matters |
|---|---|---|
| Annual AI/ML device authorizations | 295 devices | Highest annual count in the reviewed series |
| Cumulative AI/ML device authorizations | 1,451 through December 2025 | Shows the market is no longer early in volume terms |
| Dominant specialty | Radiology: 211 devices, or 71.5% | The authorization boom remains heavily imaging-centered |
| Dominant pathway | 510(k): 97% | Most products entered through substantial equivalence rather than premarket approval |
| Software classification | 62% SaMD | Most 2025 devices were software as a medical device |
| Clinical function | 63% diagnostic; 4.7% therapeutic | The authorized universe is weighted toward detection, measurement, and interpretation support |
| Predetermined Change Control Plan adoption | 10% | A minority included a pre-specified plan for certain future modifications |
| Review time | Median 142 days; 24% cleared in under 90 days | The review process is moving quickly for a substantial subset |
| Randomized clinical trial evidence | 1.6% in a study of devices through July 2023 | Clearance volume is far ahead of randomized clinical validation |
The growth curve is clean enough to be useful. FDA AI/ML device clearances rose from 6 in 2015 to 91 in 2022, then accelerated to 221 in 2023, 253 in 2024, and 295 in 2025.[1] That sequence is the strongest evidence that AI has moved from demonstration language into a measurable medical-device category.
It is also where misreadings begin. A clearance count measures regulatory throughput. It does not, by itself, measure clinical adoption, comparative effectiveness, impact on outcomes, workflow acceptance, or payer value. A device can be authorized for a narrow image-analysis task and still be described in procurement slides as part of a broad “AI transformation.” The FDA count deserves attention precisely because it is concrete; it should not be asked to prove more than it measures.
Radiology Is Still the Center of Gravity
Radiology accounted for 211 of the 295 AI/ML device clearances in 2025, or 71.5% of the total.[1] Cardiovascular devices followed at 26 clearances, or 8.8%, and neurology accounted for 14, or 4.7%.[1] Orthopedic devices represented 3.4%.[1] Product code QIH, radiological computer-assisted detection and diagnosis software, alone accounted for 75 clearances, one quarter of all 2025 clearances.[1]
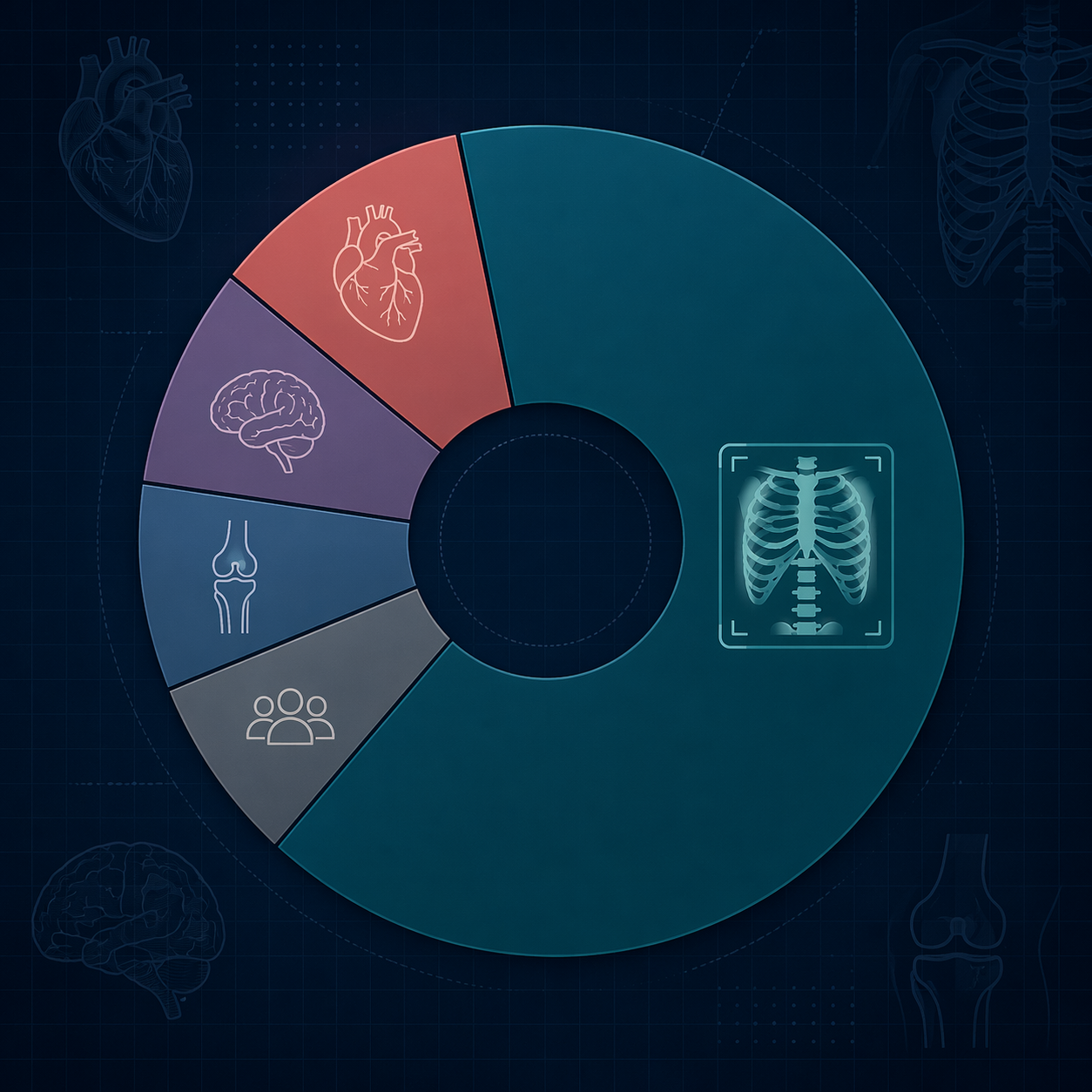
There is nothing surprising, or inherently suspect, about radiology leading. Imaging produces standardized digital inputs, lends itself to lesion detection, segmentation, triage, quantification, and quality-control tasks, and has a long history of computer-aided detection. The mistake is treating radiology’s maturity as a proxy for healthcare AI’s maturity overall.
The uneven distribution also explains why many of the most visible commercial AI categories still sit near imaging operations: detection of suspected findings, worklist prioritization, measurement support, image reconstruction, and reporting assistance. Readers looking for a company-by-use-case view of that part of the market can use this AI medical imaging companies landscape as a companion map, but the regulatory point is narrower: the FDA AI device universe is still mostly an imaging universe.
Most Devices Still Enter Through 510(k)
In 2025, 97% of AI/ML device clearances in the Innolitics dataset went through the 510(k) pathway.[1] That is the most important regulatory fact beneath the headline count. A 510(k) clearance generally rests on substantial equivalence to a predicate device; it is not the same thing as a finding that a device has independently improved patient outcomes in routine care.
This pathway pattern makes sense in a field where many products refine or extend familiar clinical tasks: identifying a finding on an image, flagging a case for review, measuring a structure, or supporting diagnostic interpretation. It also means the annual count is more incremental than revolutionary in regulatory structure. The market is growing quickly, but the dominant permission mechanism remains familiar.
The software profile is similarly concrete. Sixty-two percent of 2025 clearances were classified as software as a medical device, 63% were diagnostic, and 4.7% were therapeutic.[1] That diagnostic-versus-therapeutic split should temper broad claims about AI “treating” patients. Most authorized products are closer to detection, analysis, triage, measurement, or decision support than to autonomous therapeutic intervention.
PCCPs Are Present, but Not Yet the Default
Predetermined Change Control Plans appeared in 10% of 2025 clearances.[1] That is worth noting because AI devices can change in ways that traditional device files were not built to handle neatly. A PCCP can describe, in advance, certain planned modifications and the method for controlling them, rather than forcing every update into the same post-clearance uncertainty.
Still, 10% is not a new default. It is an early signal that lifecycle oversight is becoming more operational, not proof that the authorized AI device market has solved post-deployment change management. For readers who want the policy mechanics behind PCCPs and total product lifecycle oversight, this separate analysis of how the FDA is reshaping AI medical device regulation is the better place to go deeper.
Fast Review Does Not Equal Broad Clinical Proof
The 2025 review-time data are striking: the median review time was 142 days, and 24% of devices were cleared in under 90 days.[1] For manufacturers, that kind of timeline can shape financing, launch planning, and product-roadmap decisions. For health systems, it can create the impression that the regulatory gate has already done most of the evaluation work.
It has not. FDA authorization answers a regulatory question about whether a device may be marketed for its intended use under a particular pathway. A local evidence review still has to ask different questions: whether the device’s tested population resembles the hospital’s population, whether performance holds across scanners or sites, whether workflow burden shifts to clinicians, whether false positives create downstream costs, and whether the claimed benefit matters to the service line.
This is where “FDA-authorized” becomes a useful starting label and a poor stopping point. It tells a committee that the product has crossed a regulatory threshold. It does not tell the committee whether the device is worth buying, where it should sit in clinical workflow, or who should be accountable when the model is wrong.
The Evidence Gap Is Not a Footnote
A 2025 JAMA study examining FDA-authorized AI/ML medical devices through July 2023 found that only 1.6% cited randomized clinical trial evidence, while 46.7% of FDA summaries omitted study-design information.[3] The time window matters: the study does not directly characterize all 2024 or 2025 authorizations. But it is still too central to leave outside the interpretation of the current clearance boom.

The problem is not that every AI medical device must have an RCT before a hospital can reasonably use it. Some narrow measurement tools or workflow-support products may be evaluated appropriately through other designs, especially when the clinical risk is limited and the claim is technical. The problem is documentary: if the FDA summary does not disclose enough about study design, population, comparator, setting, and endpoints, local reviewers have to reconstruct the evidence base from fragments.
That burden lands on the CMIO, clinical engineering group, radiology quality committee, service-line lead, or value-analysis team. They are asked to translate a clearance letter into a deployment decision: turn it on for all cases, pilot it in one modality, restrict it to a subset of clinicians, require retrospective validation, monitor overrides, or decline the purchase. The thinner the public evidence record, the more that decision depends on local testing and vendor-provided material.
What the Functional Mix Says About Maturity
The broader functional taxonomy reinforces the same pattern. Singh et al. reported that 84.4% of FDA-authorized AI/ML-enabled medical devices process images.[4] They also found that quantification and feature localization declined from 81% of new devices in 2016 to 51% in 2024, while signal-based devices were rising.[4]
That does not mean image AI is stagnant. It means the authorized universe began with tasks that were especially compatible with existing digital clinical data and device-evaluation patterns. The decline in the share of quantification and localization among new devices suggests some broadening of function, but it does not erase the imaging concentration. A field can diversify and remain lopsided at the same time.
For clinical buyers, the relevant question is therefore not whether “AI” is mature. It is whether the specific task is mature: a lung nodule detection aid, a stroke triage tool, an ECG analysis model, an orthopedic planning product, or a therapeutic-support application. The FDA list is most useful when it is read at that level of granularity.
Manufacturers: A Few Familiar Leaders and a Long Tail
The 2025 manufacturer list has recognizable names at the top, but no single company defines the year. Shanghai United Imaging Healthcare had 10 clearances, Philips had 6, and Pearl Inc., Siemens, and Canon each had 5.[1] Across the broader manufacturer universe, 183 of 221 manufacturers had a single clearance.[1]
The cumulative leaderboard looks more concentrated among large imaging and device companies: GE HealthCare had 120 authorizations, Siemens Healthineers 89, Philips 50, Canon 45, United Imaging 38, and Aidoc 31.[2] That pattern fits the specialty distribution. Companies with deep imaging footprints, installed bases, and regulatory infrastructure are well positioned in a market where radiology remains the dominant category.
The long tail is just as important. A single clearance can represent a narrow but legitimate product, an early foothold, or a company that never builds a broader authorized portfolio. Procurement teams should resist reading one clearance as evidence of organizational maturity; it is evidence that one device, for one intended use, cleared one regulatory pathway.
How to Read the 1,451-Device Total
The cumulative total of 1,451 authorizations through December 2025 is a serious number.[2] It gives strategists a real market map and gives clinical leaders a way to see where FDA-reviewed AI activity has clustered. It also helps separate regulated medical-device AI from general software claims that may never come near an FDA submission.
But the total should be read with three constraints. First, it is not evenly distributed across medicine. Second, it is dominated by 510(k) clearances. Third, the public clinical evidence record, at least through the July 2023 window studied in JAMA, often lacks randomized evidence and sometimes omits basic study-design details.[1][3]
Those constraints do not make the FDA AI device list unimportant. They make it more useful. A well-read list shows which specialties have enough product density to compare options, which product codes are absorbing repeated submissions, which manufacturers have experience with the process, and where a hospital should expect to do more of its own validation work.
The Local Go/No-Go Question
For a health system, the practical decision is rarely “Should we use AI?” It is more often whether a specific authorized product should be deployed for a defined clinical task under defined monitoring conditions. The FDA record can answer some of that question, but not all of it.
- Start with the exact intended use, not the product category.
- Confirm the pathway: 510(k), De Novo, PMA, or another authorization route where applicable.
- Separate technical performance from clinical outcome claims.
- Ask whether the public summary identifies study design, population, data source, comparator, and endpoints.
- Decide what local monitoring is needed before broad deployment.
A narrow hypothetical example makes the distinction clear. An imaging AI tool may be authorized to flag a suspected finding for radiologist review. That does not automatically show that the hospital will reduce time to treatment, decrease missed diagnoses, avoid unnecessary follow-up, or improve staffing efficiency. Those are downstream claims, and they need evidence at the level of the local workflow.
That is the uncomfortable but manageable reading of 2025. The year appears to be the largest FDA AI/ML medical device year on record in this source set. The cumulative market is now large enough to count in detail. Yet the authorization boom remains narrow in specialty distribution, mostly incremental in pathway structure, and well ahead of randomized clinical validation.
References
- 2025 Year in Review: AI/ML Medical Device 510(k) Clearances, Innolitics.
- FDA AI Medical Device Tracker, IntuitionLabs.
- Clinical Validation of Artificial Intelligence and Machine Learning-Based Medical Devices Authorized by the US Food and Drug Administration, JAMA, 2025.
- Functional taxonomy of AI-enabled medical devices, npj Digital Medicine, 2025.
Comments
Join the discussion with an anonymous comment.