The uncomfortable question around FDA AI medical devices is no longer whether the market is real. It is whether the public evidence available to clinicians, purchasers, and governance committees is strong enough to support the confidence that often arrives with the phrase “FDA-cleared.” By the end of 2025, tracking counts placed cumulative FDA-authorized AI/ML medical devices above 1,400, with 295 clearances in 2025 alone. Yet in a 2025 cross-sectional study of 691 FDA-authorized AI/ML devices, 46.7% of FDA decision summaries omitted study design, 53.3% omitted sample size, fewer than 2% were supported by randomized clinical trials, and fewer than 1% reported actual patient health outcomes.[1]

Those numbers do not prove that cleared devices are unsafe. They do not prove that manufacturers submitted no useful evidence to FDA. They do show that a hospital trying to make a disciplined adoption decision may find the public record missing the first questions any clinical reviewer would normally ask: Who was studied? Against what comparator? In what setting? With what outcome? And did performance hold outside the development environment?
That distinction matters because FDA clearance and clinical confidence answer different questions. Clearance establishes that a device may be legally marketed under the applicable regulatory pathway. It does not, by itself, settle whether a specific hospital should rely on the device for triage, measurement, documentation, image interpretation, or clinical decision support in its own patients.
What the Public Evidence Usually Shows
The JAMA Health Forum analysis is useful because it reads the public clearance record the way a procurement or AI governance group has to read it: not as a theoretical regulatory file, but as the information visible to outsiders after authorization. Across 691 AI/ML medical devices, the authors found that basic methodological details were often absent from FDA decision summaries, including study design in 46.7% and sample size in 53.3%.[1]
The same study found that randomized evidence was rare. Fewer than 2% of devices were supported by randomized clinical trials, and fewer than 1% reported patient health outcomes rather than intermediate performance measures.[1] In clinical terms, that means much of the visible evidence is about whether the software can perform a task under specified testing conditions, not whether its use improves diagnosis, treatment, workflow safety, or patient outcomes after deployment.
That is not a trivial paperwork problem. If a decision summary reports a performance metric without the design, sample, setting, or validation population needed to interpret it, the number becomes hard to translate into local risk. A radiology department, for example, does not only need to know that an algorithm performed well somewhere. It needs to know whether the testing population resembles its own scanners, acquisition protocols, disease prevalence, referral patterns, and patient mix.
| Question a hospital committee needs answered | What the public record may not clearly provide |
|---|---|
| Was the device tested prospectively, retrospectively, or only on curated data? | Study design was omitted from 46.7% of FDA decision summaries in the JAMA Health Forum analysis.[1] |
| How many cases or patients were included? | Sample size was omitted from 53.3% of decision summaries.[1] |
| Did use of the device improve patient care? | Fewer than 1% of devices reported patient health outcomes.[1] |
| Was the evidence randomized? | Fewer than 2% were supported by randomized clinical trials.[1] |
The Landscape Is Mostly Image-Based and Task-Specific
The shape of the market also helps explain the evidence pattern. A 2025 npj Digital Medicine taxonomy of 1,016 FDA-authorized AI medical devices found that 84.4% used images as input, and 58% performed quantification or feature localization.[2] This is a world dominated less by general clinical reasoning systems than by tools that detect, mark, measure, segment, prioritize, or quantify a defined feature in a defined data type.
For many of these products, the immediate claim is narrow: identify a suspected finding, calculate a volume, flag an image for review, or extract a feature from a scan. Narrow claims can be clinically useful. They can remove drudgery, reduce measurement variation, and help clinicians sort large volumes of imaging data. They also make it easier for a device to be evaluated on technical or diagnostic performance rather than downstream patient outcomes.
The same taxonomy found that model characteristics and training-data composition were often undisclosed in public summaries.[2] For a hospital, those missing details are not academic curiosities. Training-data composition can affect whether a model has been exposed to relevant scanner types, acquisition protocols, disease presentations, age groups, and demographic variation. Model characteristics can affect how the device behaves when software updates arrive or when clinical practice shifts.
Counts vary across FDA, academic, and commercial tracking efforts because they use different cutoff dates and inclusion methods. FDA’s public list has historically depended on identification methods within device summaries, while independent analyses may use broader search or classification strategies. The important point is not whether one count is the definitive denominator for every analysis. It is that a large and growing device class is reaching clinical markets faster than public, decision-useful validation evidence is becoming routine.
What 510(k) Clearance Answers—and What It Does Not
The structural reason is not mysterious. Most FDA-authorized AI/ML medical devices have reached market through the 510(k) pathway; published summaries place the share at approximately 97%. That pathway is built around substantial equivalence: a manufacturer shows that its device is as safe and effective as a legally marketed predicate device for the intended use. When substantial equivalence can be demonstrated, the pathway does not routinely require a new randomized clinical trial.
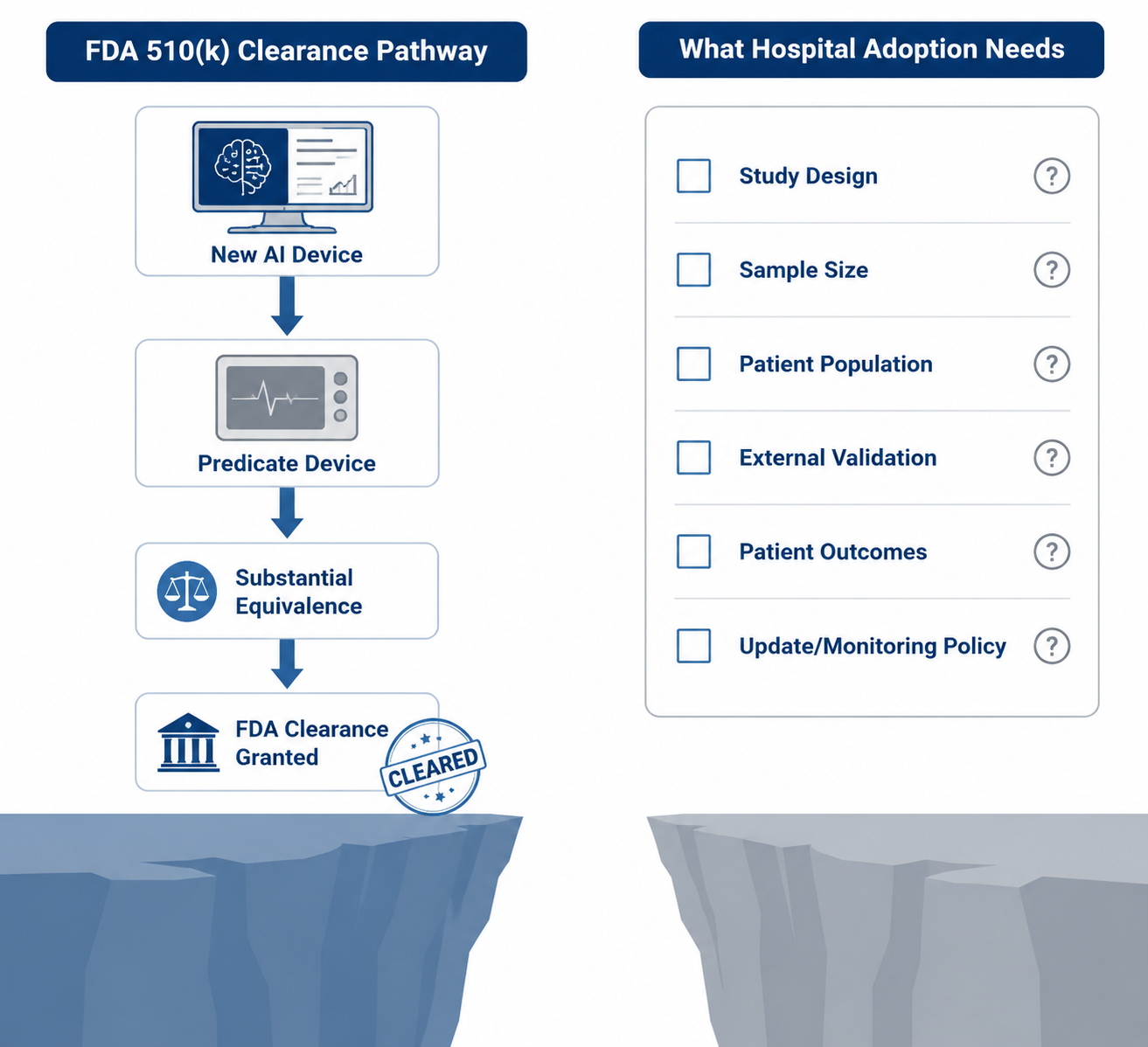
That design makes sense for many moderate-risk devices. Requiring a new outcomes trial for every incremental device change would slow access to useful technology and may add little when the device function, risk profile, and predicate comparison are well understood. But the same structure can produce a public evidence record that is thinner than what clinicians expect when a tool is being promoted as dependable clinical infrastructure.
Substantial equivalence is not the same as proof of clinical efficacy in a local workflow. A device can be cleared because it meets the regulatory standard for legal marketing, while still leaving open questions about external validation, clinical utility, equity across patient subgroups, user behavior, alert fatigue, failure modes, update governance, and post-deployment drift.
This is where vendor language often becomes too smooth. “FDA-cleared” is a meaningful statement, but it is not a substitute for the evidence packet a hospital needs before connecting a tool to patient care. The clearance may tell the committee that the device passed through a regulatory gate. It does not necessarily tell the radiologist how often a false negative occurred in a similar practice environment, the quality officer how adverse events will be detected, or the informatics lead how model changes will be communicated and validated.
The Hidden Evidence Problem
There is also a fairness issue in how the gap is described. Public evidence gaps are not the same as total evidence absence. Manufacturers may submit testing data, engineering information, performance analyses, and risk controls to FDA that are not fully visible in public summaries. Confidential commercial information, review conventions, and summary formats all shape what outsiders can see.
For hospital governance, however, invisible evidence has limited operational value. A committee cannot monitor what it cannot inspect. A clinical champion cannot defend adoption months later with data that procurement never received. A safety officer cannot design surveillance around subgroups, failure modes, or workflow hazards that were never disclosed. Confidential review may satisfy a regulatory process, but it does not automatically equip a care delivery organization to manage the device after purchase.
Why Post-Market Evidence Matters
The case for better transparency becomes stronger after clearance, not weaker. A 2025 JAMA Network Open analysis found that 5.8% of AI-enabled medical devices were ever recalled, with recalls primarily involving software bugs.[3] That figure should not be inflated into a claim that AI devices are broadly unsafe. Most recalled devices were not described as catastrophic failures, and the overall recall proportion was relatively low.
The more useful signal is the association with validation visibility. The same analysis found that devices lacking published clinical validation were at significantly higher recall risk.[3] That finding does not prove that missing publication caused the recalls. It does suggest that absence of public clinical validation may be a marker for weaker development, less mature evaluation, higher uncertainty, or simply less transparent products—each of which matters to a hospital inheriting the operational risk.
The recall analysis also reported one patient death linked to an AI device.[3] One death is not a frequency estimate for the entire category, and it should not be used to imply a widespread pattern. It is enough to remind adopters that software errors can cross the boundary from inconvenience to patient harm when they sit inside clinical workflows.
Software bugs are a particularly relevant failure mode for AI-enabled devices because implementation is not a single event. Interfaces change. Imaging protocols change. Patient populations shift. Operating systems, viewers, PACS integrations, and cloud services are updated. If a tool depends on a narrow input format or a model update changes performance, the failure may first appear as a work queue delay, an unexplained alert pattern, a missing notification, or a clinician work-around.
That is why the evidence question cannot end at authorization. A hospital adopting an AI device needs a post-market plan that is specific enough to detect degradation, not a generic assurance that the vendor monitors performance. The plan should say what will be measured locally, who reviews the signal, what threshold triggers investigation, how users report suspected failures, and what happens when the vendor ships an update.
A Practical Reading of “FDA-Cleared”
The right response is not to dismiss FDA clearance. Clearance is necessary context. It means the product has passed a federal regulatory review for the indicated use and pathway. It should be part of every procurement file, credentialing discussion, and governance review.
But it should sit at the beginning of the hospital’s evidence review, not at the end. The adoption committee still needs to ask for the materials that turn a legally marketed product into a locally governable clinical tool.
- Study design: Was validation retrospective, prospective, silent-mode, reader-study based, or interventional?
- Population and setting: Do the validation data resemble the hospital’s patients, scanners, acquisition protocols, disease prevalence, and care environment?
- External validation: Was performance tested outside the development site or vendor-controlled data source?
- Clinical outcomes: Did the study measure patient outcomes, clinician decisions, workflow effects, or only algorithm performance?
- Subgroup performance: Were relevant demographic, clinical, device, or site-level subgroups reported?
- Update policy: What changes can occur after deployment, how will users be notified, and when is local revalidation required?
- Monitoring plan: Who tracks performance, false positives, false negatives, downtime, overrides, and user-reported failures?
- Recall and safety history: Has the device, vendor, or product family had recalls, corrections, cybersecurity notices, or field safety communications?
The answers do not need to be perfect for every use case. A low-risk measurement aid used by trained clinicians may justify a different threshold than an autonomous triage tool that changes worklist priority. A documentation assistant may raise different hazards than a device that flags suspected intracranial hemorrhage. The point is to make the risk classification explicit rather than letting a clearance label flatten every device into the same adoption posture.
FDA Is Moving, but the Public Evidence Standard Is Still Unsettled
FDA has recognized that AI-enabled devices do not fit comfortably into a one-time review model. Predetermined Change Control Plans, total product lifecycle thinking, and the agency’s September 2025 request for information on real-world performance measurement all point toward a regulatory system that expects AI devices to change and to be monitored over time.
Those signals are important, but they are not the same as finalized, routine public-evidence requirements that give every hospital a usable validation dossier. Until that standard is clearer, purchasers and clinicians remain responsible for bridging the gap between legal market access and local clinical trust.
For now, the defensible posture is simple: treat FDA clearance as a regulatory milestone, then ask the clinical evidence questions anyway. If the vendor can show the study design, sample, population, external validation, outcome measures, update controls, monitoring plan, and recall history, the adoption conversation becomes specific. If those materials are missing, the hospital is not being cautious for its own sake. It is refusing to confuse authorization with assurance.
References
- Clinical Validation of Artificial Intelligence and Machine Learning-Based Medical Devices Cleared by the US Food and Drug Administration, JAMA Health Forum, 2025.
- A taxonomy of artificial intelligence medical devices approved by the US Food and Drug Administration, npj Digital Medicine, July 2025.
- Postmarket Safety Events Among FDA-Authorized AI/ML-Enabled Medical Devices, JAMA Network Open, 2025.
Comments
Join the discussion with an anonymous comment.