The short answer to the search for an FDA approved AI medical device list is this: through December 30, 2025, 1,451 AI-enabled medical devices had been FDA-authorized, with radiology accounting for 1,104 of them, or 76% of the total.[1] The language matters immediately. Most of these products are not “approved” in the strict PMA sense; about 97% reached the market through the 510(k) clearance pathway.[2]

That distinction is not regulatory pedantry. It changes what a hospital, researcher, or procurement committee can safely infer from the list. A device appearing on the FDA’s AI-enabled medical devices page means the agency has identified it as an AI- or machine learning-enabled device that has gone through an FDA marketing authorization route. It does not, by itself, establish that the product improves outcomes in a given health system, outperforms local workflow, or is ready for broad deployment without additional validation.
The FDA’s own page is still the necessary starting point, because it is the official agency reference. But it should not be treated as a complete census. The agency describes the page as a list of AI/ML-enabled medical devices identified through FDA review of device summaries and other public information, and it notes that the list is not comprehensive.[3] For a usable end-2025 picture, the FDA page has to be read alongside independent tracking by The Imaging Wire and Innolitics, both of which apply their own cutoff dates and counting methods.[1][2]
The End-2025 Count
| Measure | End-2025 figure | How to read it |
|---|---|---|
| FDA-authorized AI-enabled devices | 1,451 | Cumulative count through December 30, 2025, using end-2025 tracking from The Imaging Wire and Innolitics.[1][2] |
| Radiology devices | 1,104, or 76% | The dominant specialty category, but not the whole field.[1] |
| 2025 authorizations | 295 | A record year and a 16.6% increase over 2024’s 253.[2] |
| Main regulatory pathway | About 97% 510(k) | Mostly FDA clearance based on substantial equivalence, not PMA approval.[2] |
| 2025 SaMD share | 62% | Most 2025 clearances were pure Software as a Medical Device rather than software embedded in a device.[2] |
The most important change over the past decade is scale. AI-enabled medical device authorizations rose from 6 in 2015 to 295 in 2025.[2] That trajectory is large enough to affect capital planning, clinical governance, cybersecurity review, contracting, and post-market monitoring. It also makes sloppy language more expensive. When only a handful of products were involved, a committee could correct terms case by case. At 1,451 devices, misunderstanding the status of the list becomes an institutional workflow problem.
What “FDA Authorized” Means Here
The practical umbrella term is “FDA-authorized.” It can include devices cleared through 510(k), classified through De Novo, or approved through PMA. Those pathways are not interchangeable.
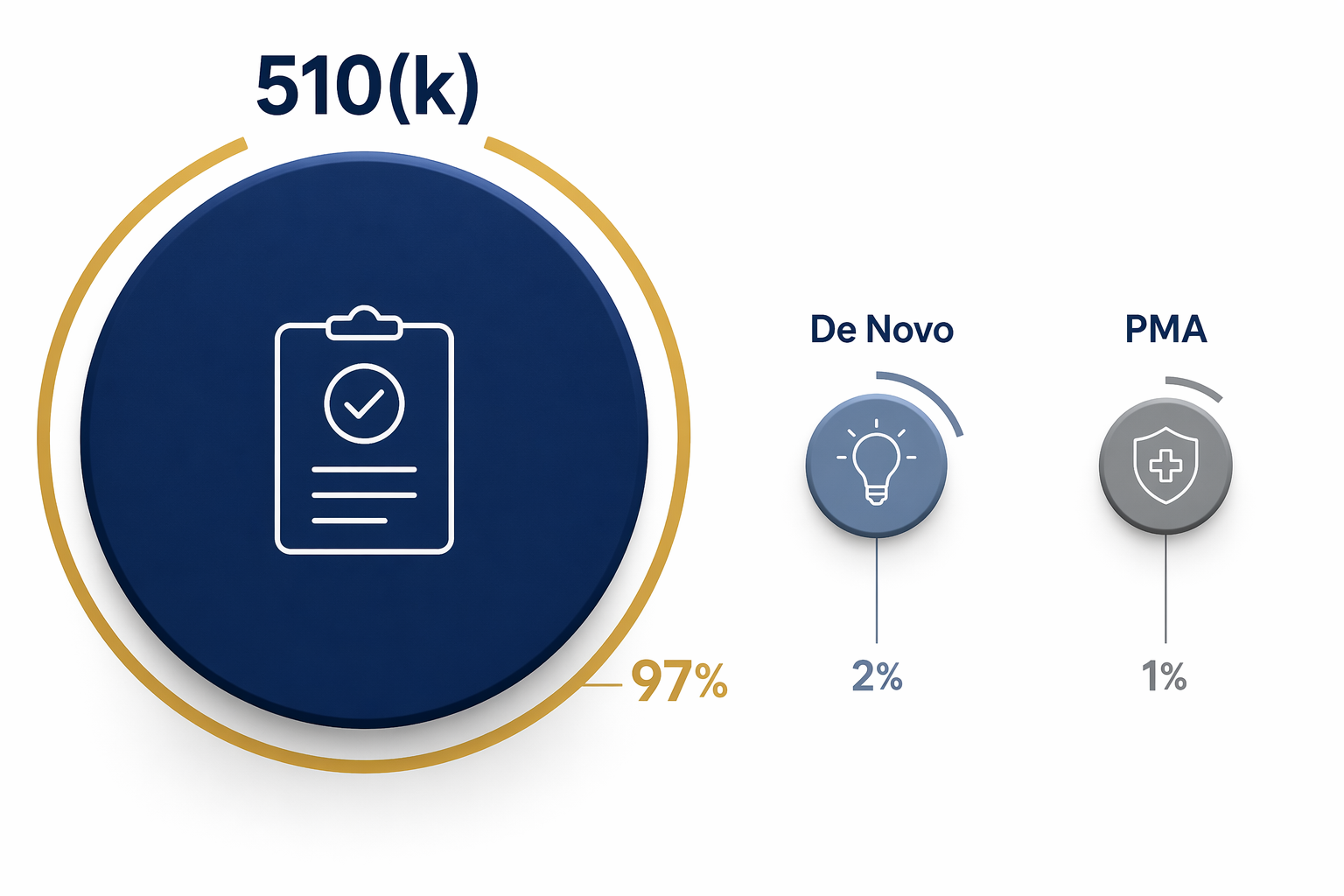
| Pathway | Share of AI device authorizations | What it supports | What it does not prove |
|---|---|---|---|
| 510(k) clearance | About 97% | The device is substantially equivalent to a legally marketed predicate for its intended use.[2] | It does not automatically prove clinical superiority, real-world effectiveness, or broad deployment readiness. |
| De Novo classification | About 2–3% | A new device type can be classified when no suitable predicate exists and general or special controls can provide reasonable assurance of safety and effectiveness.[2] | It should not be read as evidence that all later products in the category have the same clinical evidence base. |
| PMA approval | Less than 1% | The more stringent premarket approval route for certain high-risk devices.[2] | It represents only a very small portion of the AI-enabled device list. |
This is why “FDA approved” is a poor shorthand for the whole list. A 510(k)-cleared AI triage tool, a De Novo-classified device type, and a PMA-approved high-risk device may all appear in discussions of FDA-authorized AI, but they reach the market through different legal mechanisms. In procurement review, that difference affects the questions that follow: predicate comparison, intended-use language, clinical validation, monitoring obligations, integration risk, and the evidence needed before local go-live.
The 2025 median 510(k) clearance time for AI/ML devices was 142 days, according to Innolitics.[2] That number is useful for understanding review tempo. It is not a measure of how long a hospital should take to evaluate implementation, whether a model will perform on local data, or whether clinicians will act on its output.
Radiology Still Defines the List
Radiology’s 1,104 devices explain the shape of the FDA AI list more than any other fact.[1] The specialty has the imaging data, digital infrastructure, PACS-adjacent workflow, measurable detection tasks, and commercial history that made it the first large market for many AI device developers. A device can flag a suspected finding, prioritize a worklist, quantify an anatomical feature, or support image interpretation without having to replace the physician’s final judgment.
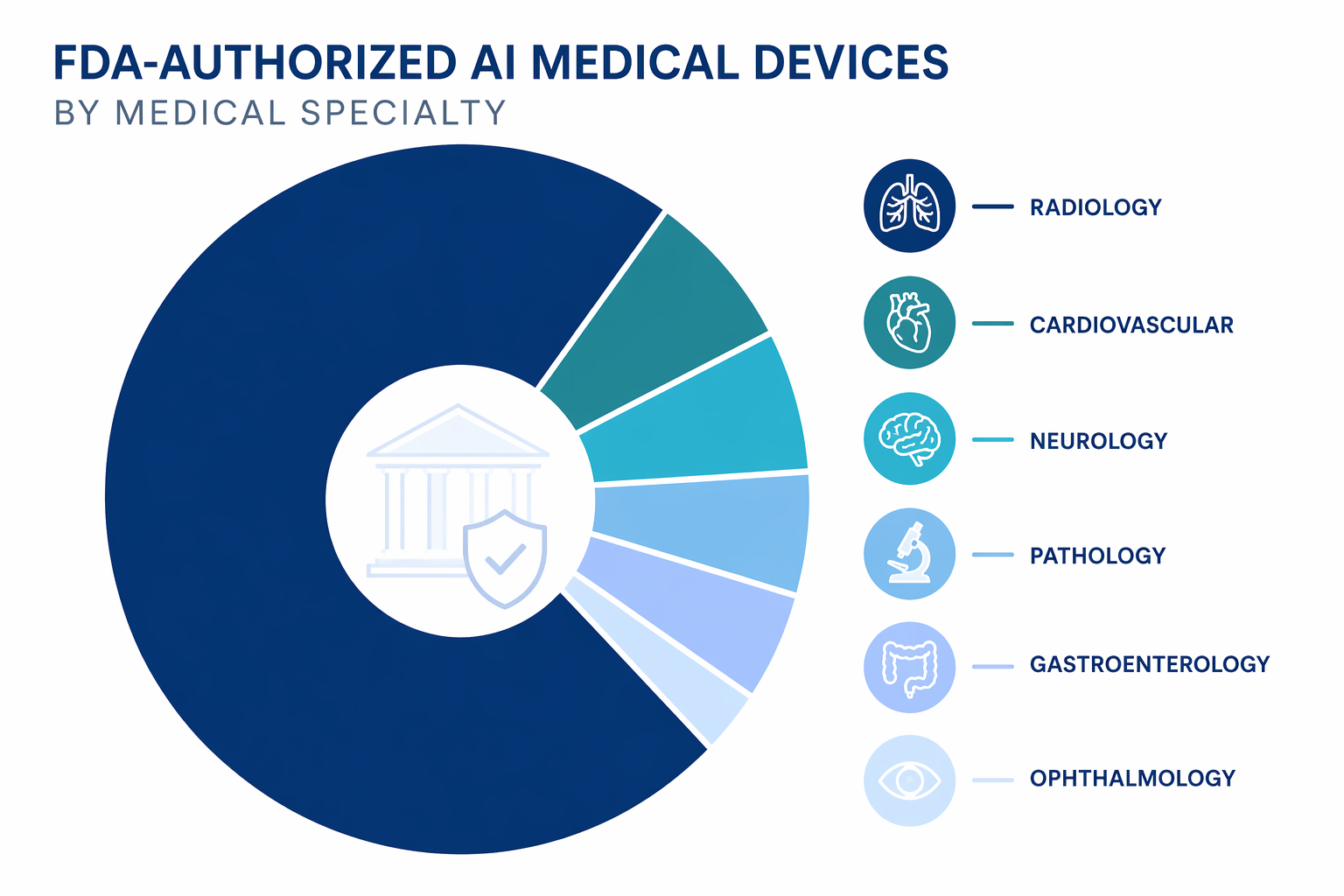
A 76% share should not be misread as proof that AI is mature across medicine. It says that FDA-authorized AI has clustered most heavily where image-based use cases and device-review precedents are strongest. For a hospital, that means the AI governance workload often begins in radiology even when the enterprise strategy is broader.
| Specialty or area | Approximate end-2025 count | Share or position |
|---|---|---|
| Radiology | 1,104 | 76% of the total.[1] |
| Cardiovascular | About 130 | Roughly 9%; the largest non-radiology category.[1] |
| Neurology | About 68 | Roughly 5%; smaller but clinically important.[1] |
| Pathology, gastroenterology, ophthalmology, and others | Smaller groups | Evidence of diversification, though far below radiology’s volume.[1] |
The non-radiology categories deserve attention precisely because they are smaller. Cardiovascular AI has moved into areas such as imaging interpretation, physiologic signal analysis, and cardiac workflow support. Neurology authorizations point toward acute detection, monitoring, and assessment use cases. Pathology, gastroenterology, and ophthalmology show how the AI device category is spreading into other visually or signal-rich specialties. The counts are still approximate because FDA panels and clinical specialty groupings do not always map cleanly onto one another.[1]
That classification problem matters in day-to-day review. A device may sit in one FDA product area while being purchased, deployed, or governed by a different clinical department. A specialty count is therefore a map, not a routing slip. The safer approach is to verify the device’s intended use, indications, target users, input data, output, and clinical environment before assigning it to a governance lane.
Who Holds the Most Clearances
The manufacturer view shows two markets at once. Large imaging and device companies hold many authorizations because AI is increasingly incorporated into existing product families. At the same time, the long tail remains substantial: Innolitics reported that 183 manufacturers had only a single clearance in 2025.[2]
| Manufacturer | Reported cumulative authorizations | Interpretation |
|---|---|---|
| GE HealthCare | 120 | The largest reported total, reflecting a broad imaging and device portfolio.[2] |
| Siemens Healthineers | 89 | Another major incumbent with repeated AI-enabled device clearances.[2] |
| Philips | 50 | A large portfolio holder across imaging and clinical technology.[2] |
| Canon | 45 | A significant imaging-market participant in the authorization data.[2] |
| United Imaging | 38 | A sizable entrant among imaging-centered manufacturers.[2] |
| Aidoc | 31 | A focused AI company with a notable acute-care and radiology footprint.[2] |
| DeepHealth | 28 | A specialized AI imaging company with multiple authorizations.[2] |
The leaderboard is useful, but it should not become a proxy for quality. A manufacturer with many clearances may have a broad, modular portfolio. A manufacturer with one clearance may have a narrow device that fits a specific clinical need. Counts do not rank clinical utility, implementation support, cybersecurity maturity, health equity performance, or post-market responsiveness.
Parent-company and subsidiary treatment can also change comparisons. Some trackers aggregate related entities; others preserve the entity named in the FDA record. For procurement and research, the record that matters is the one tied to the exact product, version, intended use, and regulatory submission under review.
What Changed in 2025
The 2025 cohort is important because it was both larger and more software-centered. Innolitics counted 295 AI/ML medical device 510(k) clearances in 2025, up 16.6% from 253 in 2024.[2] Among 2025 clearances, 62% were pure Software as a Medical Device and 63% were diagnostic in purpose.[2]
Those two figures are operationally significant. A pure SaMD product enters the hospital through a different set of questions than an AI feature embedded in a scanner or workstation. It may require separate review of cloud architecture, user provisioning, data routing, interface behavior, alert governance, model monitoring, and contractual controls. A diagnostic-purpose product also raises a different implementation burden than software that only automates measurement or administrative workflow.
Predetermined Change Control Plans also began to matter in a more visible way. Ten percent of 2025 clearances included PCCPs, according to Innolitics.[2] The FDA’s final guidance on PCCPs, issued in December 2024, explains how manufacturers can describe planned device modifications and the methods used to develop, validate, and implement those modifications within an authorized framework.[4]
PCCPs are not a permission slip for uncontrolled model drift. They are a regulated mechanism for certain planned changes. For a hospital, the relevant question is not merely whether a PCCP exists. It is what changes are covered, how the manufacturer validates them, how customers are notified, whether local configuration is affected, and whether the change alters clinical workflow.
The Foundation-Model Milestone Should Be Read Narrowly
Aidoc’s CARE1 became the first FDA-cleared foundation-model-enabled medical device in February 2025, a milestone that belongs in any serious account of the list.[2] It signals that foundation-model architectures are no longer only a research or general-purpose AI topic; at least one such device has passed through FDA review for a specified medical-device use.
The narrow reading is the responsible one. One foundation-model clearance does not mean foundation models have broadly arrived in FDA-cleared care, and it does not collapse the difference between a model architecture, a device function, an intended use, and a clinical deployment. The authorization attaches to the device and its cleared use, not to every possible downstream use of the underlying technical approach.
Evidence Quality Is a Separate Question
The FDA list is a regulatory artifact, not an outcomes registry. That is why evidence studies are useful companions to the list, but not substitutes for it. A 2025 JAMA Network Open study examined evidence characteristics for 691 FDA-authorized AI/ML-enabled medical devices, providing context on how variable the supporting evidence can be across the category.[5] The study does not turn the authorization count into an effectiveness count; it helps explain why authorization and clinical proof have to be reviewed separately.
The same caution applies to taxonomy work. A July 2025 npj Digital Medicine analysis studied 1,016 authorizations through a September 2024 cutoff and categorized devices by AI approach.[6] That kind of taxonomy helps researchers compare the field more systematically, but its cutoff is earlier than the end-2025 count of 1,451. It should be used to interpret patterns, not to override later cumulative totals.
This separation is especially important when procurement materials use the FDA list as a credential. Authorization can answer whether a device has a permitted marketed use in the United States. It does not answer whether the product improves throughput in a particular emergency department, reduces missed findings in a specific patient population, integrates safely with local reporting behavior, or maintains performance after a software update.
Why the Official FDA List Is Not the Final Count
The FDA page is authoritative for what it lists, but limited in how it is assembled. The agency identifies devices by reviewing publicly available information such as summary descriptions and labeling, using AI- and machine-learning-related terms; it states that the list is not comprehensive.[3] That means some devices with AI-like functionality may be missing if the public materials do not use the terms captured by the FDA’s process.
Independent trackers introduce their own choices. The Imaging Wire’s end-2025 analysis gives the 1,451 cumulative count and specialty distribution.[1] Innolitics provides detailed 2025 clearance statistics, manufacturer rankings, pathway mix, SaMD share, diagnostic-purpose share, PCCP adoption, and median clearance time.[2] MedTech Dive has also tracked AI-enabled device activity and market context across 2025 and 2026.[7] Differences among these sources usually come from cutoff dates, classification rules, or how entities and product variants are counted.
For institutional use, the right response is not to pick one tracker and ignore the others. Start with the FDA record for the specific product. Confirm the authorization number, pathway, decision date, intended use, product code, submitting entity, and device description. Then use independent datasets to understand category-level patterns, competitor context, and whether a vendor’s claims fit the broader market.
A Practical Reading Rule for the List
The FDA AI medical device list now describes a large, fast-growing, radiology-heavy market that is beginning to diversify into cardiovascular, neurology, pathology, gastroenterology, ophthalmology, and other areas. The 2025 record year, the rise of SaMD, early PCCP adoption, and the first foundation-model clearance all point to a more complex authorization environment.
But the list should be treated as the beginning of verification, not the final answer. It can tell a hospital which products have crossed an FDA marketing authorization threshold. It cannot, on its own, settle clinical utility, local performance, equity, cybersecurity, workflow burden, user adoption, contracting risk, or post-market governance. The more crowded the list becomes, the more valuable that distinction is.
References
- AI Medical Devices Surpass 1,400 — The Imaging Wire, March 2026.
- 2025 Year in Review: AI/ML Medical Device 510(k) Clearances — Innolitics, 2026.
- Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices — U.S. Food and Drug Administration.
- Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions — U.S. Food and Drug Administration, December 2024.
- Evidence Supporting FDA-Cleared Artificial Intelligence and Machine Learning–Enabled Medical Devices — JAMA Network Open, 2025.
- A Taxonomy of Artificial Intelligence Approaches in Medical Devices Authorized by the US Food and Drug Administration — npj Digital Medicine, July 2025.
- FDA AI-Enabled Device Tracker — MedTech Dive, 2025–2026.
Comments
Join the discussion with an anonymous comment.