The important point in the FDA's 2026 Bayesian clinical trial guidance is not that the agency has discovered Bayesian statistics. It is that CDER and CBER have now put Bayesian methods into a drug-and-biologic guidance document that explicitly reaches confirmatory settings, including primary inference in Phase III trials. The draft guidance, issued in January 2026 under a PDUFA VII commitment, is separate from the 2010 CDRH guidance for medical devices and speaks to a different review culture: one built around drug efficacy standards, substantial evidence, protocol commitments, statistical analysis plans, and submission packages that must survive advisory, supervisory, and inspection-level scrutiny. [1]
That distinction matters because the document is easy to overstate. It is a draft guidance, not final guidance. FDA guidance documents are not binding law, and even final guidance does not guarantee acceptance of a particular Bayesian design. The practical question is narrower and more useful: what does FDA now expect a sponsor to specify, simulate, justify, and verify if Bayesian evidence is going to carry confirmatory weight?

The answer begins with the document's two-pathway structure. FDA is not offering a general invitation to be flexible. It is asking sponsors to choose the inferential contract under which the trial will be judged, then to do the operational work that follows from that choice.
The regulatory change is a change in burden, not a shortcut
For sponsors working in rare disease, oncology, pediatrics, dose-finding, basket trials, subgroup borrowing, or adaptive platform trials, the appeal is obvious. Conventional randomized designs can become strained when the eligible population is small, the control arm is hard to enroll, prior information is clinically relevant, or the trial is meant to adapt as information accumulates. The draft guidance recognizes Bayesian methods in exactly those kinds of settings, including borrowing historical or external data, using nonconcurrent controls in platform trials, pediatric extrapolation aligned with ICH E11A, and adaptive designs aligned with draft ICH E20. [1][3]
Recognition, though, is not the same as deference. The guidance makes Bayesian methods more available in drug and biologic development, but it also makes the review questions more explicit. Who selected the prior? What data went into it? How relevant and recent were those data? What happens if the current trial conflicts with the prior? What operating characteristics does the design have under the null? What sensitivity analyses were prespecified? Which software generated the posterior probabilities, and how was it verified?
Those questions are not philosophical. They decide what appears in the protocol, the SAP, the Type C or CID meeting package, the clinical study report, and the eventual submission. A Bayesian design that looks elegant in a simulation deck but cannot be explained in those artifacts is not yet a regulatory design.
Two pathways, two different review conversations
Section IV.A is the load-bearing part of the draft guidance. It separates Bayesian trial designs into two broad regulatory approaches: trials calibrated to control frequentist type 1 error, and trials evaluated through a purely Bayesian decision analysis using agreed priors and utility functions. [1]
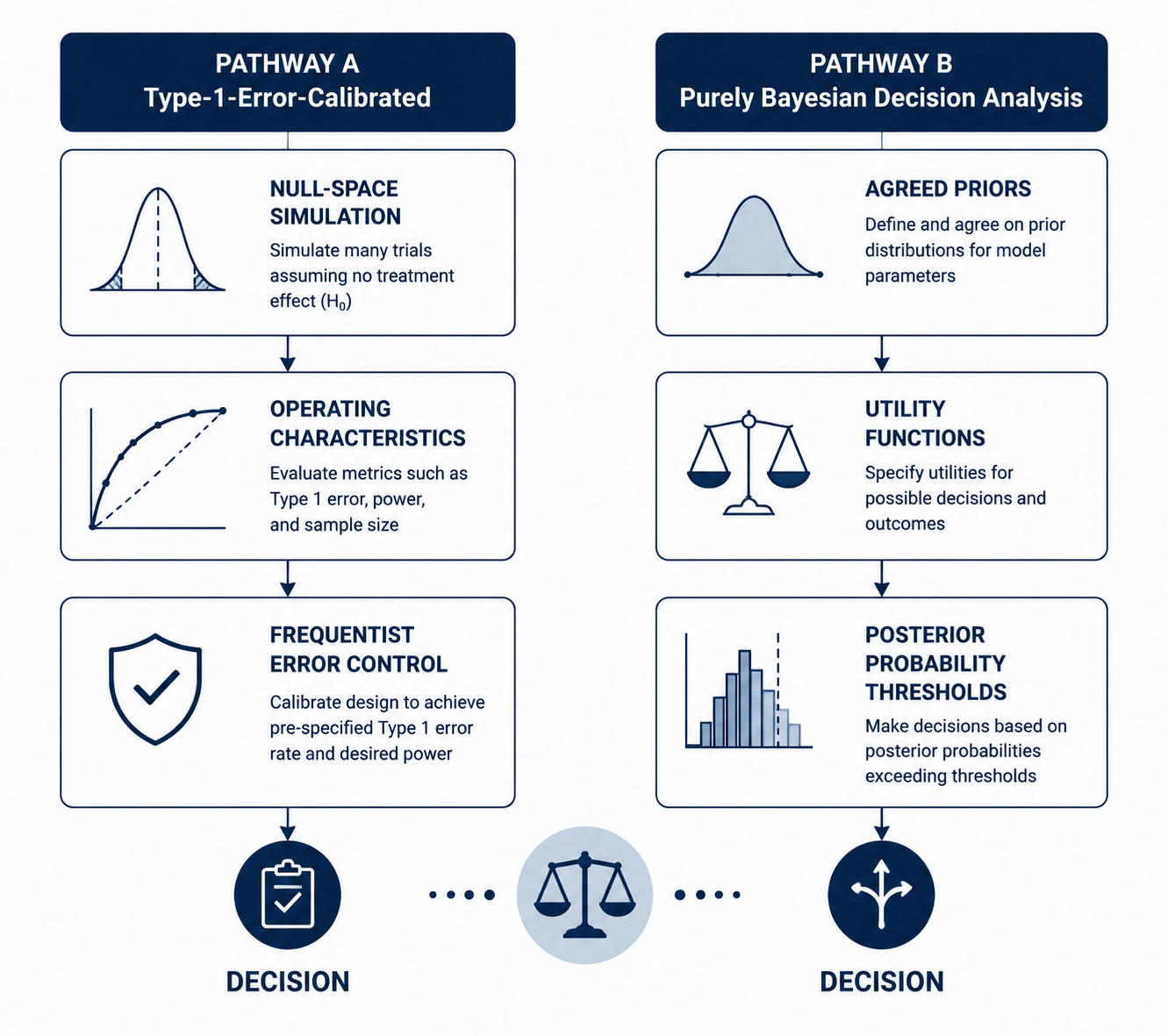
| Pathway | What FDA is asking the sponsor to defend | Where the pressure falls |
|---|---|---|
| Type-1-error-calibrated Bayesian design | The Bayesian decision rule is calibrated through null-space simulation to control false-positive risk. | Simulation assumptions, operating characteristics, thresholds, borrowing behavior, and sensitivity analyses. |
| Purely Bayesian decision analysis | The decision framework relies on agreed priors and utility functions rather than conventional type 1 error control. | Prior agreement, utility specification, decision consequences, and whether reviewers accept the proposed framework. |
The first pathway is likely to feel more familiar to many drug-development teams. A sponsor may use Bayesian machinery for posterior probabilities, adaptive borrowing, predictive probabilities, or interim decisions, but the design is still calibrated against frequentist operating characteristics. FDA expects simulation over the null space, not merely one convenient null scenario. The question becomes whether the design keeps the false-positive risk under control across the relevant combinations of nuisance parameters, accrual patterns, borrowing assumptions, missingness assumptions, and decision thresholds.
That is where the word "Bayesian" stops being a label and becomes a simulation program. If a platform trial borrows from nonconcurrent controls, the reviewer needs to see how often the design would incorrectly conclude benefit when the investigational therapy is ineffective and historical controls differ from concurrent controls. If a rare disease trial uses external natural history data, the reviewer needs to see what happens when the external data are less exchangeable than hoped. If a basket trial shares information across subgroups, the reviewer needs to see when borrowing helps a small subgroup and when it inappropriately imports evidence from a different disease context.
The second pathway is more ambitious and probably more difficult to operationalize in a confirmatory drug setting. A purely Bayesian decision analysis asks FDA and the sponsor to agree not only on the prior distributions, but also on the utility functions that map trial outcomes to regulatory decisions. Berry Consultants' January 2026 analysis correctly treated this as a place where interpretation may diverge: the distinction between the guidance's type-1-error-calibrated pathway and the purely Bayesian pathway is consequential, but not always effortless to locate in real designs that mix Bayesian posterior rules with frequentist calibration. [4]
A sponsor should not assume that using posterior probabilities automatically places the trial in the purely Bayesian lane. Nor should it assume that a Bayesian adaptive feature becomes easy to defend simply because type 1 error is simulated somewhere in the appendix. The pathway choice needs to be visible early, because it determines what FDA is being asked to accept: a calibrated decision rule with Bayesian components, or a Bayesian decision system whose priors and utilities are themselves part of the evidentiary agreement.
Why the pathway choice changes rare disease, oncology, and platform trial planning
The near-term effect will probably be most visible in programs that already have a reason to reach beyond a conventional fixed randomized design. In rare diseases and pediatrics, prior information may come from natural history studies, adult efficacy data, previous trials, registry evidence, or mechanistic expectations. In oncology, Bayesian designs may support dose-finding, subgroup borrowing, basket structures, or adaptive allocation. In platform trials, the attraction is often the possibility of borrowing from nonconcurrent controls or updating decisions as multiple therapies enter and leave the platform. [1][3]
None of those applications becomes presumptively valid under the draft guidance. The framework instead gives sponsors a way to state the regulatory problem precisely. If the design is meant to preserve conventional error control, the sponsor must show the design's behavior under null and alternative scenarios. If the design is meant to make a purely Bayesian decision, the sponsor must secure agreement on the prior and utility structure before the trial depends on it.
This is also why the guidance may matter beyond programs that ultimately use Bayesian primary inference. It gives review divisions, sponsors, and statisticians a common vocabulary for deciding when borrowing is credible, when prior information is too fragile, and when an adaptive rule needs more simulation before it belongs in a pivotal protocol.
Prior construction is where the submission work begins
The guidance's treatment of priors is one of its most practical contributions. A prior is not a decorative input if it can move the posterior probability that supports approval. It is part of the evidence-generating system, and FDA expects the sponsor to say how it was constructed before the current data are known. [1]
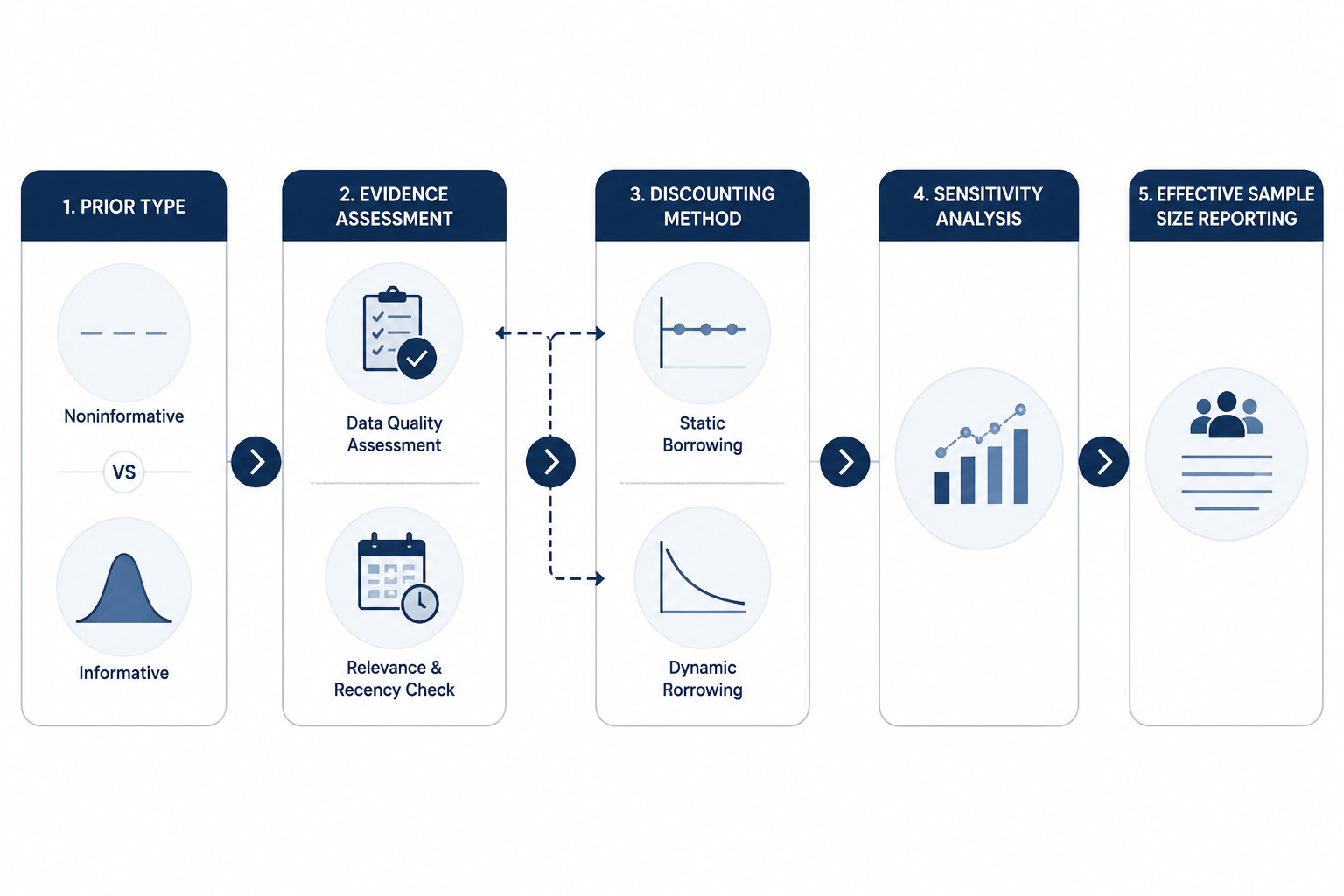
Noninformative priors require less justification because they are not intended to contribute substantive outside information. They are not risk-free. Berry Consultants points to edge cases such as Beta(ε,ε) priors that can produce spurious certainty in all-events or no-events data patterns. [4] That kind of example is a useful reminder that "noninformative" is not a regulatory spell. The sponsor still needs to show that the prior behaves reasonably in the data configurations the trial may actually generate.
Informative priors require much more. The sponsor must justify the quality, relevance, and recency of the source data; explain whether the prior data are clinically and operationally exchangeable with the proposed trial; and predefine how conflict between prior information and current trial data will be handled. The FDA draft guidance places that work in Section V, and specialist analyses have emphasized that this is likely to become one of the most scrutinized parts of a Bayesian submission. [1][4][5]
A historical control dataset, for example, is not automatically relevant because it measures the same endpoint. The trial population may have shifted. The diagnostic pathway may have changed. Supportive care may have improved. Endpoint ascertainment may differ. Missing data may have been handled differently. A pediatric extrapolation prior may be scientifically reasonable, but the bridge from adult data to pediatric inference still needs a stated biological and clinical rationale.
Static discounting and dynamic borrowing are not interchangeable
Section V.D draws attention to discounting methods. Static discounting assigns a fixed weight to prior or external information. Dynamic borrowing lets the amount of borrowing depend on agreement between the prior information and the accumulating current data. The second approach is attractive because it appears to protect the trial when the prior conflicts with the observed data. It can do that, but the guidance and outside analyses warn that dynamic borrowing can also produce pathological behavior, including cases where weaker observed data paradoxically support stronger conclusions. [4][5]
That warning deserves more attention than it often receives. Dynamic borrowing is not a reviewer reassurance by itself. The sponsor has to show how the borrowing rule behaves across plausible and implausible levels of prior-data agreement, and not only in the scenario where the historical data happen to be correct. The more consequential the borrowing is for the primary endpoint, the less persuasive it is to present one favorable simulation and a sentence about adaptivity.
Effective sample size reporting belongs in the same conversation. For dynamic borrowing methods, Berry Consultants emphasizes that effective sample size should be reported as a function of observed data rather than as a single fixed number. [4] That is not a formatting preference. If the amount of borrowed information changes with the trial result, then a single headline effective sample size can conceal the very behavior the reviewer needs to understand.
The protocol and SAP have to carry the design, not merely describe it
Section VIII turns the statistical concept into submission mechanics. The protocol should identify the Bayesian features that affect trial conduct or interpretation. The SAP should specify the model, priors, decision criteria, sensitivity analyses, simulation plan, and computational methods. For analyses relying on Markov chain Monte Carlo, the sponsor should address convergence diagnostics. The submission should also include software verification sufficient for the role the software plays in the analysis. [1]
- The protocol should make clear which trial decisions depend on Bayesian calculations, including interim adaptations, borrowing rules, stopping criteria, and primary inference.
- The SAP should prespecify priors, posterior decision thresholds, simulation assumptions, operating characteristics, sensitivity analyses, and handling of prior-data conflict.
- The simulation report should include null-space simulations when type 1 error control is part of the regulatory claim.
- The computational package should document MCMC convergence diagnostics when MCMC is used and provide software verification appropriate to the analysis.
- The submission should explain effective sample size, especially when dynamic borrowing makes the borrowed information depend on observed data.
Sensitivity analysis is not a late-stage comfort exercise in this setting. If a design uses an informative prior, the sensitivity analyses should examine design priors and alternative assumptions that matter to the regulatory conclusion. If the result only survives under the sponsor's preferred prior and collapses under a reasonable skeptical prior, that is not a nuisance finding; it is part of the evidence.
Berry Consultants also notes ambiguity around the placement of skeptical and enthusiastic priors within the guidance's framework, as well as an arguably insufficient emphasis on predictive probabilities. [4] Those are not academic quibbles. Predictive probabilities often drive interim decisions in adaptive trials, and skeptical or enthusiastic priors can determine how robust a design appears before enrollment begins. Sponsors should assume that these choices may need to be defended in reviewer meetings even if the final guidance text later clarifies the taxonomy.
Comment timing and draft status should be verified from primary records
As of July 2026, the guidance remains a draft. The Federal Register notice is the primary record to use for procedural details, including the public comment deadline. Available sources show a discrepancy between a March 13, 2026 deadline in the Federal Register record and a later April 30, 2026 date reported by a secondary source; that discrepancy should be resolved against the Federal Register before relying on a specific deadline. [2]
That may sound administrative, but it reflects the larger caution around the document. Draft guidance can change. Review divisions may apply it unevenly while the field adjusts. A sponsor designing a pivotal Bayesian trial in 2026 should treat the draft as highly relevant evidence of FDA thinking, not as a guarantee that any particular prior, borrowing rule, or decision threshold will be acceptable.
Early engagement is not optional for the hard cases
The guidance lands in an FDA environment that already has channels for discussing complex trial designs. FDA's Center for Clinical Trial Innovation, established in 2024, and the Complex Innovative Trial Design Meeting Program offer routes for sponsors to bring Bayesian designs forward before the protocol is locked. [6][7] Those meetings matter most when the design is doing something reviewers cannot evaluate from a conventional alpha-controlled template.
A useful early meeting package should not ask FDA whether it is generally open to Bayesian methods. The draft guidance has already answered that at a policy level. The better package asks more concrete questions: whether the proposed pathway is the right one, whether the prior sources are acceptable candidates, whether the null-space simulations cover the right scenarios, whether the dynamic borrowing rule has been stress-tested adequately, and whether the planned sensitivity analyses will address the review division's concerns.
Implementation will also depend on staffing. Applied Clinical Trials and PharmaVoice both identified practical barriers, including the need for Bayesian-trained biostatisticians and cultural adjustment across sponsors and CROs. [6][7] That constraint should temper any prediction that the guidance will rapidly convert mainstream confirmatory development. The review framework may now be clearer than the workforce pipeline.
What sponsors should prepare now
For a sponsor considering Bayesian primary inference or meaningful Bayesian borrowing, the first task is classification. Decide whether the trial is intended to be type-1-error-calibrated or presented as a purely Bayesian decision analysis. Ambiguity at this level will later appear as ambiguity in the SAP, simulation report, and reviewer briefing book.
The second task is prior governance. Identify the prior sources, document why they are relevant, evaluate their quality and recency, and define how conflict with the current data will be handled. If dynamic borrowing is planned, describe the borrowing rule in a way that can be simulated, challenged, and reproduced. If a noninformative prior is planned, confirm that it does not create unreasonable behavior in edge-case data patterns.
The third task is simulation discipline. A simulation plan that only demonstrates attractive performance under the sponsor's expected treatment effect is incomplete. For a calibrated design, null-space simulation is central. For borrowing designs, conflict scenarios are central. For adaptive platform trials, nonconcurrent-control assumptions and time trends need attention. The simulation package should show how the design fails, not only how it succeeds.
The fourth task is computational readiness. MCMC diagnostics, reproducible code, version-controlled simulation programs, software verification, and traceable outputs should be planned before the submission clock starts. A posterior probability in a clinical study report is only as reviewable as the computational path that produced it.
The January 2026 draft guidance legitimizes Bayesian confirmatory designs for drugs and biologics in a way that prior FDA device guidance did not. It also raises the evidentiary burden for transparency, simulation, prior justification, and computational documentation. Whether it becomes transformative will depend on final guidance language and reviewer practice. For now, sponsors have enough to start building Bayesian trial strategies that can be reviewed as regulatory evidence rather than admired as statistical innovation.
References
- Bayesian Methodology in Clinical Trials for Drugs and Biological Products, U.S. Food and Drug Administration, January 2026.
- Bayesian Methodology in Clinical Trials for Drugs and Biological Products; Draft Guidance for Industry; Availability, Federal Register, January 2026.
- FDA Issues Draft Guidance on Bayesian Methodology in Clinical Trials for Drugs and Biological Products, Alston & Bird, January 2026.
- A Guide to the FDA Bayesian Guidance, Berry Consultants, January 30, 2026.
- FDA's Bayesian Guidance: Implications for Drug and Biologic Development, Frontiers in Medicine, April 14, 2026.
- FDA Draft Guidance Opens Door for Bayesian Methods in Clinical Trials, Applied Clinical Trials, January 14, 2026.
- How FDA's Bayesian Guidance Could Change Clinical Trial Design, PharmaVoice, February 10, 2026.
Comments
Join the discussion with an anonymous comment.