
By the end of 2025, the U.S. Food and Drug Administration had authorized 1,451 AI- and machine learning-enabled medical devices, with a record 295 clearances issued in 2025 alone. Radiology accounts for approximately 76% of all authorized devices (1,104 devices), followed by cardiology at roughly 9% and neurology at about 5%. These figures, drawn from the most recent comprehensive tracking by The Imaging Wire and analyzed through March 2026, represent a nearly 26% increase over the prior year's cumulative total. The pace of regulatory authorization has accelerated to the point where a clinician or health system procurement team can no longer evaluate these tools on a case-by-case basis without a structured framework for assessing evidence quality.
This article provides an integrated, evidence-grounded review of machine learning in medical diagnosis for healthcare professionals and clinical researchers. It examines four interconnected domains: the diagnostic accuracy reported in controlled research settings, the quality of evidence underlying FDA authorizations, the documented gap between laboratory performance and real-world deployment outcomes, and the evolving regulatory frameworks that will shape future adoption. The core thesis is straightforward: ML models demonstrate high diagnostic accuracy in curated research environments — with pooled area under the curve (AUC) values ranging from 0.87 to 1.0 across major specialties — but significant evidence gaps, methodological heterogeneity, and deployment challenges mean that real-world performance frequently diverges from published results. Fewer than 2% of FDA-authorized AI devices are supported by randomized clinical trial evidence.
Diagnostic Accuracy by Specialty: What the Meta-Analyses Show
The most comprehensive meta-analysis of deep learning diagnostic performance in medical imaging remains Aggarwal et al. (2021), published in npj Digital Medicine. The review examined 503 studies across multiple specialties and reported pooled AUC ranges that appear impressive at first glance.

| Specialty | Clinical Application | Pooled AUC Range | Studies Using Prospective Data |
|---|---|---|---|
| Ophthalmology | Diabetic retinopathy, age-related macular degeneration, glaucoma (retinal fundus photos, OCT) | 0.933 – 1.00 | 8 of 503 total |
| Respiratory Imaging | Lung nodule detection, lung cancer classification (chest X-ray, CT) | 0.864 – 0.979 | 2 of 503 total |
| Breast Imaging | Breast cancer detection (mammogram, ultrasound, MRI, digital breast tomosynthesis) | 0.868 – 0.909 | 0 of 503 total |
These AUC values are frequently cited by vendors and proponents as evidence that ML diagnostic tools are ready for widespread clinical deployment. However, the meta-analysis itself documents several critical limitations that temper this conclusion. Heterogeneity between studies was high, with extensive variation in methodology, terminology, and outcome measures. No studies provided a prespecified sample size calculation. The majority of studies were classified as high risk of bias under the QUADAS-2 tool, a standardized instrument for assessing diagnostic accuracy studies. Only 8 ophthalmology studies, 2 respiratory imaging studies, and 0 breast imaging studies used prospectively collected data. In other words, the evidence base is dominated by retrospective, single-center, often small-sample studies that are methodologically prone to overestimating diagnostic performance.
The Singh et al. 2025 taxonomy of 1,016 FDA authorizations provides additional context on what these devices actually do. Among devices classified as performing data analysis — which accounts for 85.6% of all authorized devices — 65% perform quantification or feature localization, 12.9% perform triage, 7.2% perform diagnosis, 6.9% perform detection, 6.1% perform combined detection and diagnosis, and 1.7% perform prediction. The majority of authorized devices are therefore assistive tools that flag or measure findings for human review, not autonomous diagnostic systems. No evidence of large language models (LLMs) in the authorized device list was found as of the study's September 2024 cutoff.
The Evidence Quality Gap: What FDA Summaries Reveal
If the meta-analytic evidence for diagnostic accuracy is weaker than commonly assumed, the evidence base underlying FDA authorizations is weaker still. A 2025 study published in JAMA examined the FDA summaries of 691 AI-enabled medical devices and found that 46.7% of summaries did not describe the study design used to support the clearance, and 53.3% omitted the sample size. Only 6 devices (1.6%) cited a randomized controlled trial, and fewer than 1% (3 devices) reported actual patient health outcomes. These findings are consistent with a separate analysis of 903 FDA-approved AI devices published in JAMA Network Open in 2025, which found that clinical performance studies were reported for only about half of the devices.

The explanation for these gaps lies in the regulatory pathway. Nearly all cleared AI devices — approximately 97% — entered the market via the 510(k) pathway, which requires demonstration of substantial equivalence to a predicate device already on the market, rather than de novo clinical validation of safety and efficacy. This means that a device can be cleared without any new clinical data if the manufacturer can argue it is similar enough to a previously cleared device. The predicate device itself may have been cleared through the same mechanism, creating a chain of devices that have never been prospectively validated in a clinical trial. Over a median follow-up of 4.5 years, 40 devices (5.8%) were subject to 113 recall actions, primarily due to software bugs. Only 5.2% of devices had adverse events reported post-clearance.
Real-World Performance: When Lab Results Don't Translate
The gap between controlled-study performance and real-world deployment is perhaps best illustrated by the case of the Epic Sepsis Model (ESM). Widely deployed across U.S. hospitals as a clinical decision support tool for early sepsis detection, the model was found in post-deployment analyses to have missed two-thirds of sepsis cases while frequently issuing false alarms. The model's AUC, sensitivity, and specificity were significantly worse after deployment relative to initial reported performance. This case is not an isolated anomaly but rather a documented example of a broader pattern: ML diagnostic tools that perform well on curated, retrospective datasets often degrade when exposed to the messiness of real clinical environments.
Several factors drive this lab-to-clinic gap. Equipment variation — different scanner manufacturers, imaging protocols, and software versions — can alter the input data distribution in ways that degrade model performance. Population differences between the training cohort and the deployment site — in age distribution, comorbidity burden, disease severity, and demographic composition — can shift the relationship between features and outcomes. Workflow integration failures, such as alert fatigue from high false-positive rates or clinician distrust of model recommendations, can render even an accurate model ineffective in practice. And model drift — the gradual degradation of predictive performance as the underlying data distribution changes over time — means that a model validated at deployment may be substantially less accurate months or years later.
For a detailed critical appraisal of the Epic Sepsis Model's updated validation, including the Wong et al. 2026 multicenter prospective study, see our Epic Sepsis Model v2 evidence appraisal.
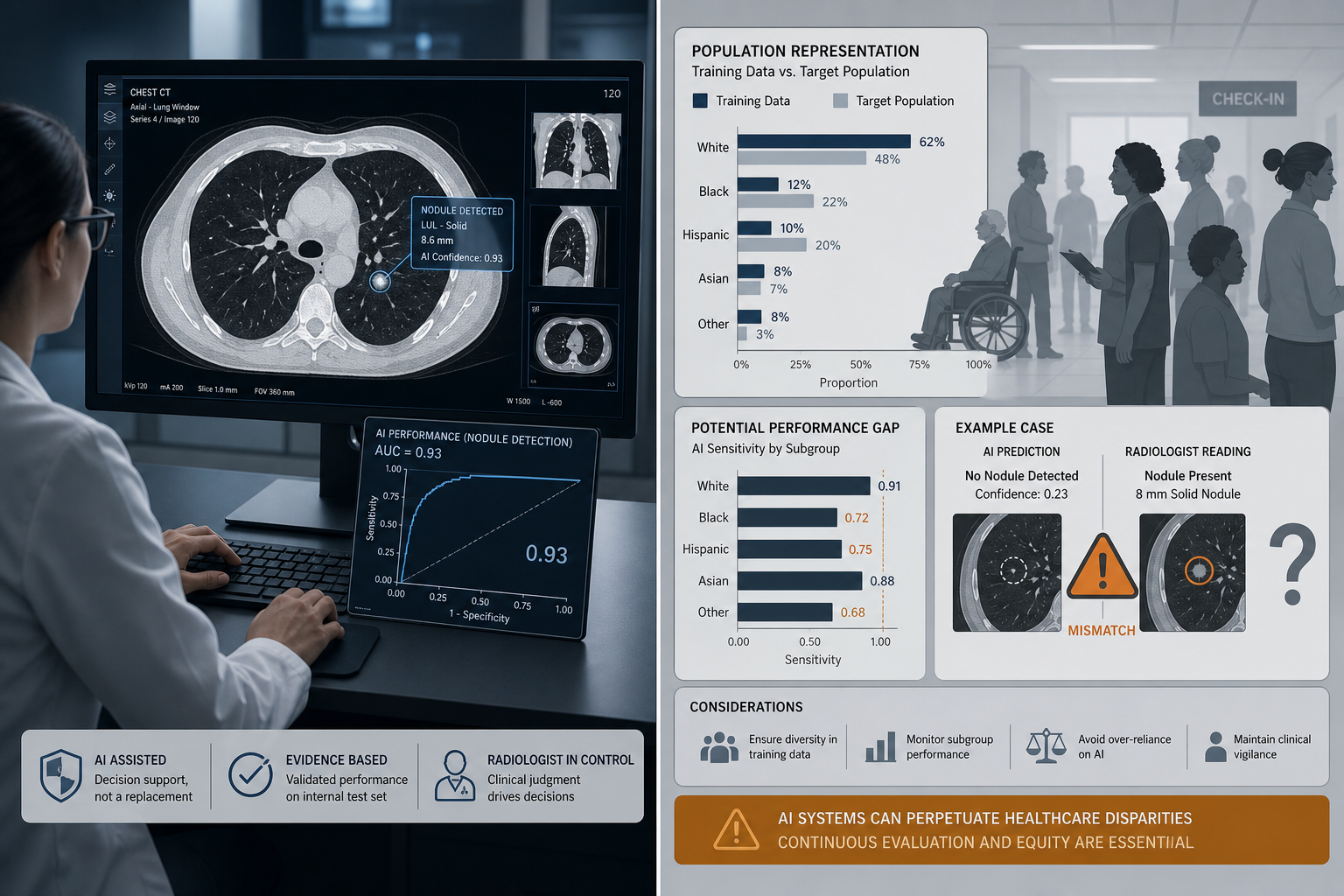
Bias and Generalizability: Who Is Missing From the Data?
The generalizability of ML diagnostic tools depends critically on whether the training data and validation populations reflect the diversity of the patients who will ultimately be assessed by the tool. The evidence suggests that current devices fall short of this standard. The 2025 JAMA Network Open study of 903 FDA-approved AI devices found that fewer than one-third of devices reported sex-specific performance data, and only one-quarter addressed age-related subgroups. This means that for the majority of authorized devices, clinicians have no way of knowing whether the tool performs equally well for male and female patients, or for pediatric, adult, and geriatric populations.
The consequences of this data gap are not theoretical. Dermatological AI systems have demonstrated lower diagnostic accuracy for melanoma in darker-skinned individuals, a direct result of training predominantly on fair-skinned images from patients in the United States, Europe, and Australia. A widely used hospital resource allocation algorithm was found to systematically under-allocate care to Black patients because it used healthcare spending as a proxy for health need — spending that is itself shaped by historical inequities in access to care. In the MIMIC-III critical care database, imbalanced representation of racial groups yielded recall rates as low as 25% for in-hospital mortality prediction in certain populations.
- Less than one-third of FDA-cleared AI devices report sex-specific performance data.
- Only one-quarter of devices address age-related subgroup performance.
- Dermatology AI underperforms on darker skin tones due to training dataset composition.
- Hospital resource allocation algorithms have demonstrated racial bias when using proxy variables correlated with structural inequities.
- Over half of published clinical AI models use data exclusively from the United States or China, limiting global generalizability.
For a deeper examination of bias taxonomy, audit frameworks, and mitigation strategies, see our dedicated article on algorithmic bias and health equity in clinical AI.
Regulatory Evolution: From 510(k) Dominance to Lifecycle Management
The regulatory framework for AI/ML medical devices is undergoing a significant transformation, driven by recognition that the traditional 510(k) pathway — designed for hardware devices with fixed specifications — is poorly suited to software that can change its behavior through learning. The FDA has taken several concrete steps toward a lifecycle management approach.
| Regulatory Development | Date | Key Provisions |
|---|---|---|
| Finalized Predetermined Change Control Plan (PCCP) Guidance | December 2024 | Establishes framework for manufacturers to specify in advance how an AI/ML device may be modified through learning while maintaining safety and effectiveness |
| Draft Total Product Lifecycle (TPLC) Guidance | January 2025 | Proposes expanded post-market surveillance requirements and continuous performance monitoring for AI/ML devices |
| PCCP Adoption in New Clearances | 2025 | 10% of AI/ML device clearances in 2025 included PCCPs, signaling early but growing adoption of the framework |
The PCCP framework is particularly significant because it attempts to solve the fundamental regulatory challenge posed by continuously learning algorithms. Under the traditional model, any modification to a cleared device — including retraining on new data — requires a new 510(k) submission. This creates a disincentive for manufacturers to improve their models post-market and leaves deployed devices operating on potentially outdated training data. The PCCP allows manufacturers to specify in advance the types of changes that will be made, the methodology for validating those changes, and the performance thresholds that must be maintained. The FDA finalized the PCCP guidance in December 2024, and 10% of 2025 clearances already included PCCPs.
The draft TPLC guidance, released in January 2025, extends this lifecycle approach by proposing expanded post-market surveillance requirements. These include continuous performance monitoring, regular reporting of real-world performance data, and mechanisms for identifying and addressing model drift. For a detailed explanation of model drift detection, retraining governance, and the regulatory framework around continuous learning algorithms, see our model drift in clinical AI article.
Internationally, the European Union's AI Act will impose high-risk obligations on medical AI systems, with compliance deadlines beginning in 2026–2027. The Act classifies medical devices that incorporate AI as high-risk systems subject to requirements for risk management, data governance, transparency, human oversight, and accuracy. The convergence of the FDA's lifecycle approach and the EU's risk-based framework suggests that the regulatory environment for ML diagnostic tools will become substantially more demanding over the next three to five years.
Implications for Clinical Adoption and Future Research
The evidence reviewed in this article supports neither wholesale adoption of ML diagnostic tools nor their blanket rejection. Rather, it points to the need for a structured, evidence-critical evaluation framework that accounts for the specific strengths and documented limitations of each tool in its intended deployment context.
For researchers, the evidence gaps identified in this review point to several priorities. Prospective validation studies with prespecified sample size calculations and diverse, well-characterized populations are urgently needed. Standardized reporting guidelines — such as CONSORT-AI for clinical trials and STARD-AI for diagnostic accuracy studies — should be adopted as a condition of publication. Post-market surveillance studies that track real-world performance, including subgroup analyses by sex, age, race, and ethnicity, are essential for understanding generalizability. And the regulatory shift toward lifecycle management, including PCCPs and TPLC requirements, should be supported and informed by independent research on model drift, retraining governance, and the clinical impact of algorithmic changes over time.
For practical context on how AI clinical decision support tools are being deployed in real-world primary care settings, including the challenges of workflow integration and clinician adoption, see our AI clinical decision support in primary care article.
The trajectory of ML in medical diagnosis is not predetermined. The technology has demonstrated genuine capability in controlled settings, and the regulatory infrastructure is evolving to address the unique challenges of continuously learning algorithms. But the evidence base today is thinner than the volume of FDA clearances might suggest. The path from high pooled AUCs in retrospective meta-analyses to reliable, equitable diagnostic tools in everyday clinical practice runs through prospective validation, transparent reporting, diverse population data, and lifecycle regulatory oversight. These are not optional additions to the development pipeline — they are prerequisites for responsible clinical deployment.
Comments
Join the discussion with an anonymous comment.