FDA’s January 2025 draft guidance on AI-enabled medical devices was issued under docket FDA-2024-D-4488, with public comments closed April 7, 2025; as of July 2026, it remains draft guidance rather than final FDA policy.[1] That status matters. It means manufacturers should not treat every sentence as settled law. It also means teams building AI-enabled device software functions now have a clear view of where FDA is trying to move the evidentiary burden: transparency and bias are not clean-up work for the labeling writer after design freeze.
The practical surprise is how early these topics land. A transparency claim can become a user-interface requirement. A fairness promise can become a validation-dataset variable. A model card can expose whether the submission summary is supported by design evidence or padded with marketing language. The draft guidance was announced by FDA as part of a broader effort to provide recommendations for lifecycle management and marketing submissions for AI-enabled medical devices.[2] For development teams, the useful reading is not “what should we say?” but “which design control file, validation protocol, human factors plan, or clinical study budget must change?”
| FDA draft expectation | Where it lands in development work | What should be traceable by submission time |
|---|---|---|
| Use a user-centered approach to transparency | User needs, UI requirements, labeling strategy, training materials, human factors plan | Evidence that intended users receive and understand the information needed for safe and effective use |
| Validate explainability information in its use context | Interface design, clinical workflow analysis, usability testing, risk controls | Evidence that explanations help rather than mislead users in the intended environment |
| Address AI bias as a systematic risk | Risk management file, data management plan, validation protocol, subgroup analysis plan | Rationale and evidence showing performance across relevant groups and conditions |
| Analyze subgroup performance | Validation dataset design, case report forms, site contracts, statistical analysis plan | Performance results by sex, age, race, ethnicity, disease variables, clinical site, acquisition equipment, and device configurations |
| Use U.S.-relevant clinical validation evidence | Site selection, study budget, OUS data justification, enrollment planning | Clinical validation evidence that reflects U.S. patients, practice patterns, and device use conditions |
| Provide model card information | Submission summary, public-facing communication, postmarket lifecycle documentation | A concise description of model purpose, data, performance, limitations, and update controls grounded in the actual evidence package |
Transparency Starts Before the Label Exists
Appendix B is easy to misread if a team is looking only for labeling obligations. It describes transparency through a user-centered communication framework: identify intended users, understand the use environment, determine what information those users need, decide how and when that information should be communicated, and evaluate whether the communication works.[1] That sequence belongs in design planning, not in the final labeling sprint.
The first engineering consequence is that “intended user” has to be more specific than a regulatory noun. A radiologist, an emergency physician, a medical assistant acquiring an image, a patient using a home-monitoring app, and an administrator configuring thresholds do not need the same transparency content. Some need limitations at the point of interpretation. Some need acquisition warnings before the algorithm runs. Some need configuration constraints, not model architecture detail.
FDA’s draft also gives transparency a business-case rationale that teams can use internally: Appendix B states that transparency has been shown to “more than double willingness to use a device.”[1] That sentence should not be inflated into proof that every explanation improves performance or adoption. It supports a narrower point: FDA sees transparent communication as materially relevant to whether users will appropriately accept and use AI-enabled devices.
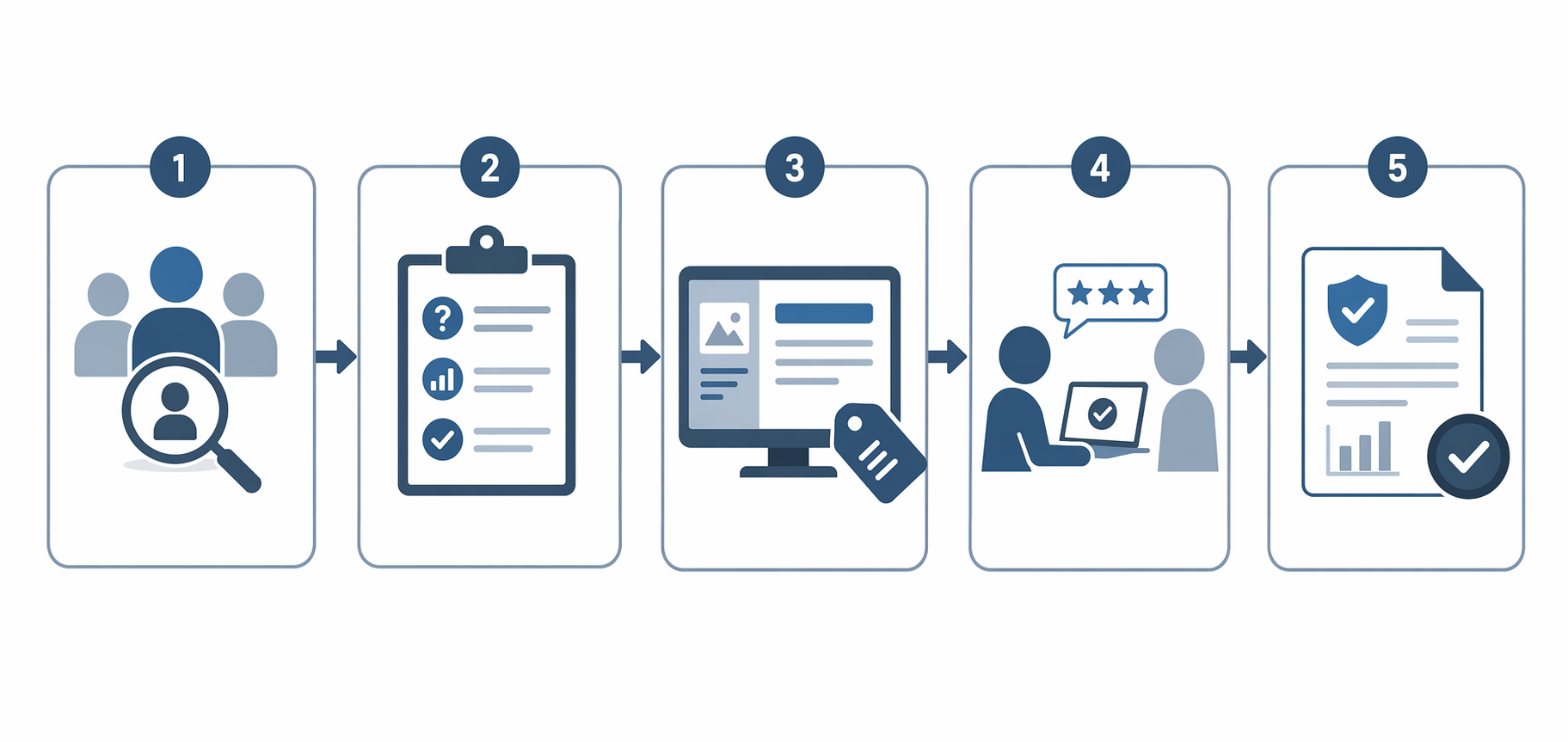
A workable transparency workflow looks like this: define the user and environment, translate user needs into interface and labeling requirements, build the explanation into the workflow, validate comprehension, then carry the evidence into the submission and model card. The weak version is familiar: a polished PDF appendix appears after the UI is locked, and the usability team is asked to prove that clinicians understand a warning they never saw during formative testing.
- User definition: identify who acts on the AI output, who configures the device, who reviews exceptions, and who bears the consequence of a wrong or misunderstood output.
- Information need: decide what the user must know about intended use, input requirements, output meaning, limitations, confidence or uncertainty information, and appropriate follow-up.
- Interface placement: determine whether the information belongs in the primary screen, an alert, an acquisition prompt, a report, training, labeling, or a model card.
- Validation method: test whether the intended user can interpret and act on the information under the expected use conditions.
- Submission trace: connect transparency content back to risk controls, design inputs, usability evidence, and performance limitations.
This is where transparency becomes expensive if delayed. If a device output includes a heat map, uncertainty band, confidence score, triage category, or “reason for alert,” that element is not merely educational content. It can change user behavior. Appendix D warns that explainability tools not validated for their specific use context “could significantly mislead users,” and the draft treats those tools as subject to human factors and usability expectations.[1] A visual explanation that looks convincing but has not been evaluated with the intended user population can become a risk control with no proof that it controls the risk.
For an engineering lead, the practical move is to separate three things that often get bundled together: the model’s internal reasoning, the explanation shown to the user, and the user action expected after seeing it. FDA does not need a philosophical account of intelligence in the interface. It needs evidence that the information presented to the user supports safe and effective use of the device. That evidence may come from formative studies, summative usability testing, labeling comprehension work, simulated-use testing, or other human factors methods appropriate to the device and pathway.
Readers who need the broader cross-jurisdictional context for explainability can compare this FDA approach with other regimes in explainable AI medical device requirements. The narrower point here is more operational: if an explanation appears in the workflow, the design file should show why it is there, what user need it addresses, what risk it controls, and how the team tested comprehension or actionability.
Bias Becomes a Validation Planning Problem
The draft guidance uses a systems-risk definition of AI bias: the “potential tendency to produce incorrect results in a systematic, but sometimes unforeseeable way.”[1] That is not the same thing as a full academic taxonomy of fairness metrics. FDA’s framing is tied to device risk, performance, intended use, and the possibility that systematic error may harm one group or use condition more than another.
That distinction is useful because it keeps the work auditable. A team can debate equalized odds, calibration, representativeness, or health equity impacts for a long time. Those debates may be necessary for some devices, and foundational concepts are better handled in an algorithmic bias glossary. The FDA submission problem is more immediate: did the manufacturer plan, collect, analyze, and explain evidence sufficient to show that the device performs appropriately across the relevant patients, sites, equipment, and configurations?
The subgroup list in the draft is not vague. FDA identifies analysis across sex, age, race, ethnicity, disease variables, clinical data site, data acquisition equipment, and device configurations.[1] Those variables touch different owners inside a company. Race and ethnicity may affect case report forms and consent workflows. Acquisition equipment may affect site qualification and metadata capture. Device configuration may affect software version control and test-environment records. Disease variables may affect both inclusion criteria and the statistical analysis plan.
| Subgroup dimension | Planning implication before validation starts |
|---|---|
| Sex and age | Ensure enrollment, data abstraction, and analysis plans can support performance estimates or justified limitations for relevant patient groups. |
| Race and ethnicity | Capture variables early enough and consistently enough that the final dataset does not leave the team explaining missing fields after lock. |
| Disease variables | Define clinically meaningful disease states, severities, or comorbidities that could affect model performance. |
| Clinical data site | Track site-level performance so results are not silently dominated by one institution or workflow. |
| Data acquisition equipment | Record scanner, sensor, imaging protocol, device model, or other acquisition variables that could change inputs. |
| Device configurations | Preserve configuration and software-version traceability for the model, preprocessing pipeline, thresholds, and operating modes. |
The uncomfortable part is that subgroup analysis cannot be made credible by writing a better paragraph at submission time. If the validation dataset does not contain the variable, the analysis cannot be performed. If a subgroup is too small, the confidence around the estimate may be too wide to support much. If acquisition equipment was not tracked, a site effect may be confused with a hardware effect. If disease severity was captured inconsistently, a performance gap may be unexplainable even when it is clinically important.
That is why bias planning should sit inside the validation protocol and statistical analysis plan, not only inside the risk-management narrative. At protocol stage, the team should decide which subgroup variables are required, how they will be captured, which analyses are primary or exploratory, how missingness will be handled, what minimum descriptive reporting is expected, and when a performance difference becomes a risk-management issue rather than a footnote.
For imaging AI, this can become particularly concrete: equipment vendor, scanner model, imaging protocol, reconstruction settings, acquisition site, and patient demographics may all matter. A separate imaging AI bias evaluation protocol can help teams think through those mechanics. The FDA draft’s contribution is to make the issue a submission-readiness question: can the manufacturer show that subgroup performance was anticipated, measured, and interpreted as part of device validation?
U.S.-Relevant Validation Evidence Changes the Study Budget
The draft guidance also has a location problem that can be easy to underestimate. It recommends that clinical validation data come from at least three geographically diverse U.S. sites.[1] That expectation is not just a regulatory preference; it affects site contracting, data-use agreements, IRB timing, monitoring, sample availability, and budget.
The guidance does not ban outside-U.S. validation data. It does, however, warn that AI-enabled devices may require a higher proportion of U.S. data in clinical validation than traditional devices, depending on the device and the relevance of the OUS data to U.S. clinical practice.[1] That is a higher evidentiary bar than some teams will have assumed if their model was trained or first validated in a health system with different workflows, population mix, imaging equipment, lab methods, or practice patterns.
The practical answer is not automatically “run every study only in the United States.” The answer is to justify data relevance before the validation plan is locked. If OUS data are included, the submission should be able to explain why those data represent the intended U.S. use population and environment, where they differ, and how the remaining U.S. validation evidence addresses the gap. A late justification written after the dataset is assembled is weaker than a protocol that anticipated the question.
Model Cards Should Not Be Marketing One-Pagers
Appendices E and F provide model card templates for AI-enabled device software functions.[1] Their existence is a signal: FDA expects certain information about the model, data, performance, limitations, and lifecycle controls to be organized in a form that can support public-facing transparency. That does not make the model card a substitute for the full submission. It makes it a compressed view of evidence that should already exist elsewhere.
A strong model card should be drafted from the design history and validation package, not from the launch narrative. If the model card says the device is intended for a particular population, the validation evidence should support that population. If it describes limitations, those limitations should map to risk analysis, labeling, and user-interface controls. If it reports subgroup performance, the numbers should trace to the locked validation dataset and statistical analysis plan.
For a broader framework discussion, including how FDA-style model cards relate to CHAI, DIHI, and other clinical AI transparency formats, see the clinical AI model card comparison. The submission-specific point is simpler: a model card created after the evidence package is finished will reveal gaps; a model card drafted early can help identify them while there is still time to change the study plan.
What Should Move Upstream Now
The draft guidance sits within a larger set of submission expectations for AI-enabled device software functions. Teams that need the complete domain-by-domain view should use the full FDA AI-enabled device software functions draft guidance breakdown. For transparency and bias, the immediate development impact is concentrated in a smaller set of artifacts.
- Design inputs should specify what transparency information each intended user needs and where that information appears in the workflow.
- Risk management files should treat misleading, misunderstood, or overtrusted explanations as hazards or contributing factors where appropriate.
- Human factors plans should include explainability elements, warnings, limitations, confidence displays, and other AI-specific communication features when they affect user action.
- Validation protocols should require capture of subgroup variables, site information, acquisition equipment, and device configuration metadata before the dataset is locked.
- Statistical analysis plans should predefine how subgroup performance will be reported, interpreted, and escalated if differences appear.
- Model card drafts should be versioned with the evidence package so public-facing transparency remains consistent with validated claims and known limitations.
Requirements will still vary by device type, risk profile, intended users, data modality, and submission pathway. A locked diagnostic algorithm, an adaptive imaging workflow tool, and a patient-facing triage application will not need identical transparency content or identical subgroup analysis. The useful discipline is to make the rationale explicit: why these users, why these variables, why this validation design, why this explanation, and why this level of U.S. clinical evidence.
Lifecycle planning also matters. FDA’s AI device policy direction has increasingly emphasized total product lifecycle management, including change control and transparency around model updates. For teams working through predetermined change control plans and related lifecycle questions, the broader PCCP, TPLC, and transparency discussion is the better place to connect this draft guidance to the larger regulatory arc.
The Compliance Boundary
The draft guidance does not turn transparency into a generic virtue statement or bias into a one-line assurance that the model was trained responsibly. It points to evidence FDA can ask to see: user-centered communication decisions, usability validation for explainability tools, subgroup performance analysis, U.S.-relevant clinical validation, and model card content that reflects the actual device evidence package.
Manufacturers that treat transparency and bias as submission-writing tasks will be exposed late in development, when the missing pieces are expensive: unplanned usability testing, incomplete race and ethnicity fields, untracked acquisition equipment, subgroup analyses with too little data, or a model card that says more than the validation package can support. Teams that convert the draft guidance into design inputs, validation protocol requirements, statistical analysis plans, and traceable lifecycle evidence will be better positioned if FDA’s final guidance preserves the same direction.
The caution is straightforward: this remains draft guidance as of July 2026. Before locking a submission strategy, manufacturers should check FDA’s final version when published and reconcile any changes to the transparency appendices, model card templates, subgroup expectations, and clinical validation recommendations.
Comments
Join the discussion with an anonymous comment.