The Evidence Gap Problem: What the Data Shows About FDA AI Device Summaries
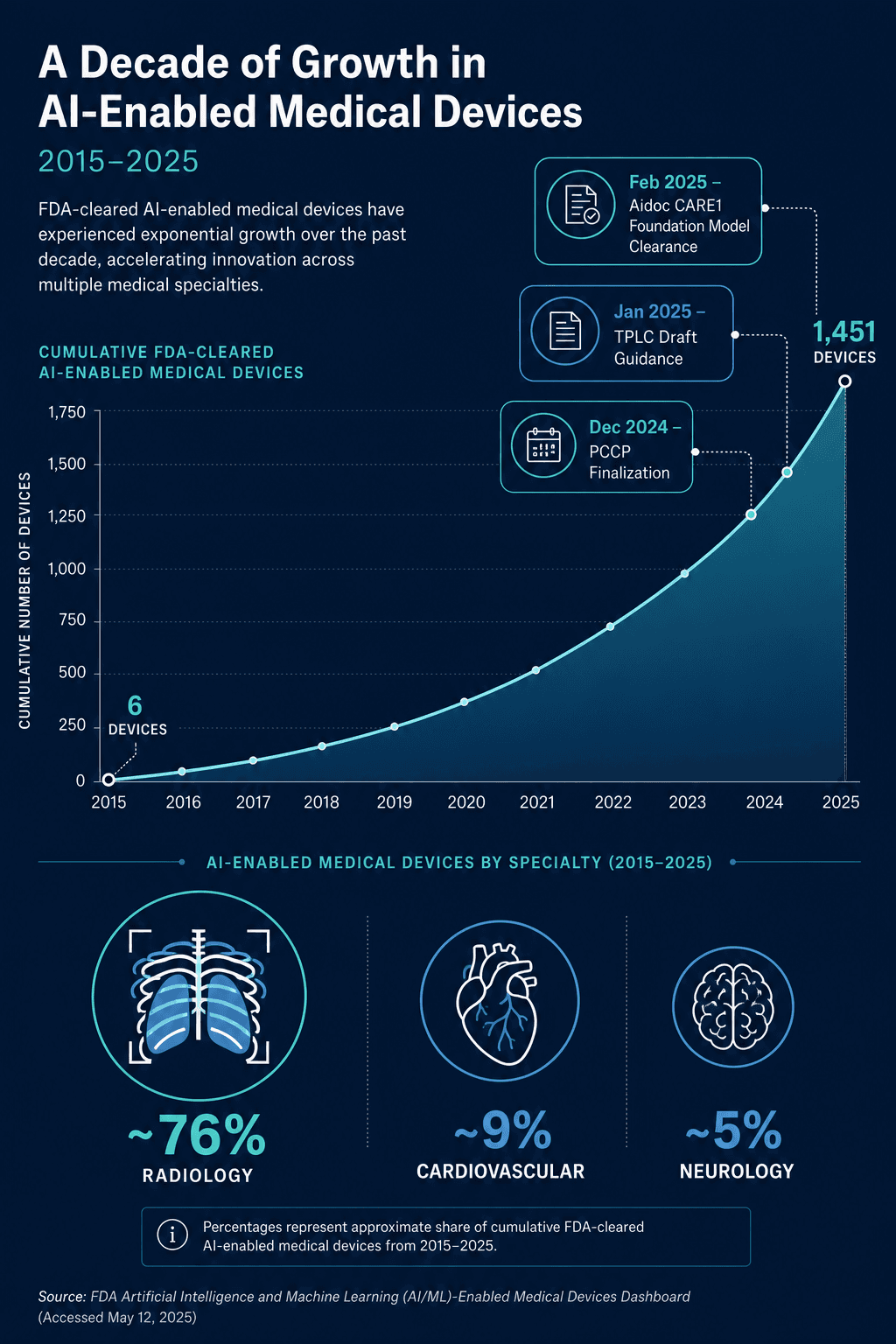
The FDA has authorized a cumulative total of 1,451 AI/ML-enabled medical devices as of the end of 2025, a figure that has grown from just 6 devices in 2015 to a record 295 clearances in 2025 alone. This rapid expansion, however, has not been matched by a corresponding increase in the quality of clinical evidence underpinning these authorizations. A 2025 cross-sectional study published in JAMA analyzed 691 FDA-cleared AI devices authorized through July 2023 and found that less than 2% were supported by randomized clinical trials. More troubling for regulatory affairs professionals: 46.7% of the FDA 510(k) summaries reviewed did not describe the study design used to validate the device, and 53.3% omitted the sample size entirely.
These transparency deficits are not merely academic concerns. They create real operational risk for health systems evaluating AI tools for procurement, for clinicians trying to assess whether a device will perform reliably in their patient population, and for manufacturers who must navigate an increasingly demanding regulatory environment. The same JAMA study found that only 5.2% of authorized AI devices had any adverse event reports on file, and 5.8% were ever recalled — mostly due to software bugs rather than clinical performance failures. The low adverse event reporting rate may reflect underreporting rather than genuine safety, given that post-market surveillance infrastructure for AI devices remains immature.
The implications for manufacturers are direct and consequential. A 510(k) submission that omits study design or sample size information may have passed FDA review under earlier standards, but the agency is now signaling clearly that such omissions will no longer be acceptable. The FDA's response to these documented evidence gaps takes the form of three interconnected regulatory modernization tools: the Total Product Life Cycle (TPLC) framework, Predetermined Change Control Plans (PCCPs), and new transparency and labeling requirements. Each tool addresses a specific dimension of the evidence problem, and together they represent a fundamental shift from a pre-market review paradigm to continuous lifecycle oversight.
The Total Product Life Cycle (TPLC) Framework: January 2025 Draft Guidance
On January 7, 2025, the FDA issued draft guidance titled Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations (docket number FDA-2024-D-4488). This document represents the agency's most comprehensive statement to date on how it expects manufacturers to manage AI-enabled devices across the entire product lifecycle — not just at the point of initial market authorization.
The guidance identifies nine documentation areas that the FDA recommends manufacturers address in their marketing submissions. These are not optional checkboxes; they form the basis for what the agency considers a complete and defensible submission for an AI-enabled device.
| Documentation Area | What It Covers |
|---|---|
| Device Description | Intended use, indications for use, device overview, and AI-specific functionality description |
| User Interface | Design considerations, human factors testing, and how AI outputs are presented to users |
| Labeling | Instructions for use, warnings, precautions, and AI-specific disclosure statements |
| Risk Assessment | Hazard identification, risk analysis, and risk control measures specific to AI behavior |
| Data Management | Data sources, collection methods, labeling processes, and data quality assurance |
| Model Description and Development | Algorithm architecture, training methodology, hyperparameters, and training data characteristics |
| Validation | Verification and validation activities, including test datasets, metrics, and acceptance criteria |
| Device Performance Monitoring | Post-market monitoring plan, performance metrics, and processes for detecting performance degradation |
| Cybersecurity | Security risk management, vulnerability assessment, and update mechanisms |
What distinguishes the TPLC framework from earlier FDA guidance is its emphasis on continuous lifecycle management rather than a single pre-market review event. The guidance explicitly addresses the reality that AI models can change over time — through retraining, fine-tuning, or deployment in new clinical contexts — and that these changes require ongoing oversight. The framework also introduces specific recommendations for bias mitigation, including testing device performance across demographic subgroups defined by age, sex, race, and ethnicity.
For regulatory affairs teams, the TPLC guidance creates immediate documentation obligations. Even if a device was cleared before January 2025, the guidance signals what the FDA will expect in future submissions and, increasingly, in post-market reviews. Manufacturers should begin auditing their existing device documentation against the nine TPLC areas to identify gaps before they become compliance issues.
Predetermined Change Control Plans (PCCPs): Enabling Iterative Improvement
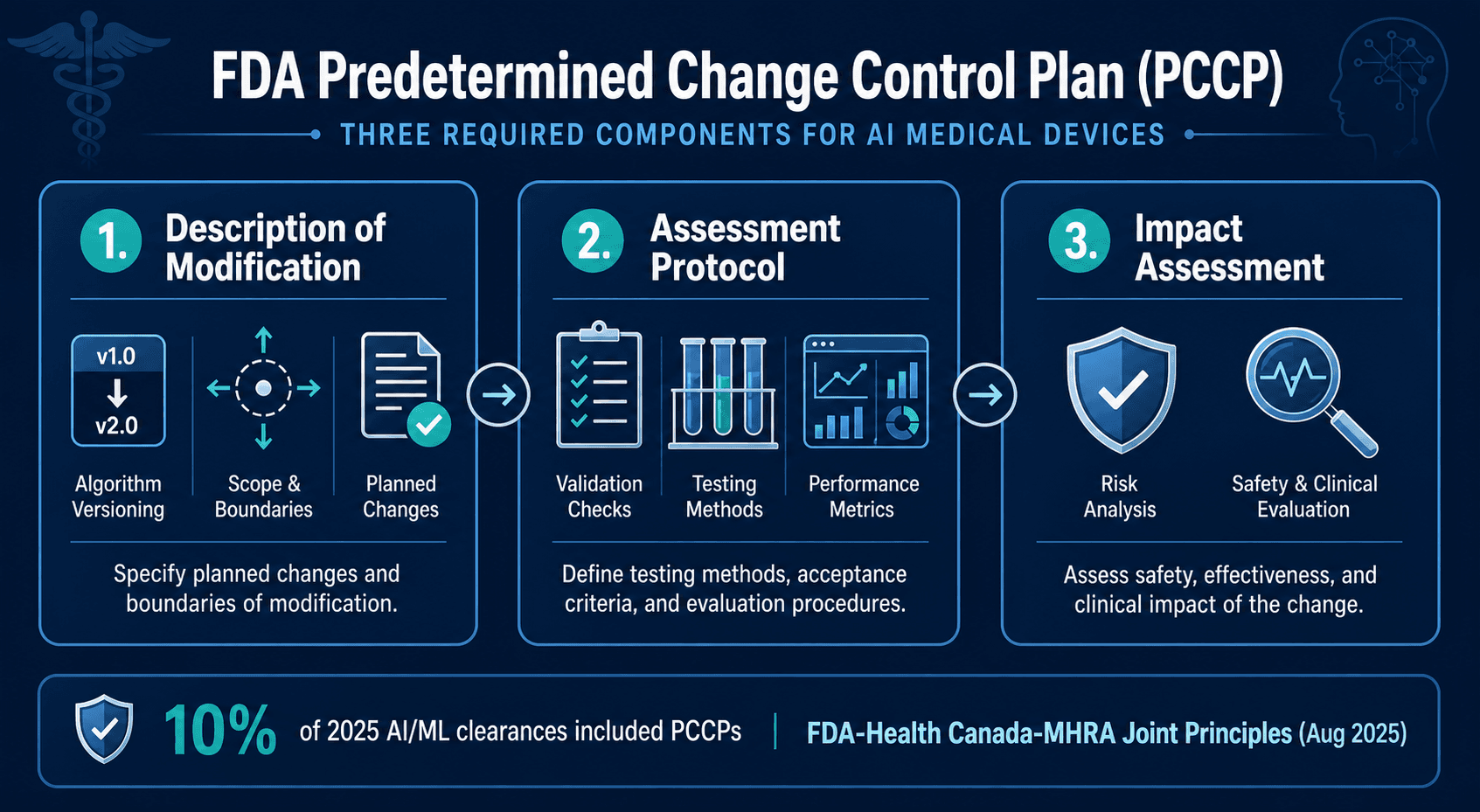
The Predetermined Change Control Plan (PCCP) is arguably the most consequential regulatory innovation in the FDA's AI device toolkit. Finalized in December 2024, the PCCP framework allows manufacturers to pre-specify planned modifications to an AI-enabled device — including algorithm updates, retraining protocols, and performance threshold adjustments — and receive FDA authorization for those changes without submitting a new 510(k) or De Novo request each time.
A PCCP must include three required components:
- Description of Modification: A detailed specification of the types of changes the manufacturer intends to make, including the scope, frequency, and triggers for each modification. This section must be precise enough that the FDA can evaluate the potential impact of changes that have not yet occurred.
- Assessment Protocol: The methodology the manufacturer will use to validate and verify each modification before implementation. This includes pre-defined performance metrics, acceptance criteria, test datasets, and validation procedures.
- Impact Assessment: An analysis of how each type of modification could affect device safety and effectiveness, including risk assessment updates and any necessary labeling changes.
Adoption of PCCPs has been measured but meaningful. In 2025, 10% of all AI/ML device clearances included a PCCP — a notable figure for a framework that was only finalized in December 2024. This adoption rate suggests that early-adopter manufacturers, particularly those with mature quality management systems, are already operationalizing the PCCP pathway.
A landmark case study is Aidoc's CARE1 foundation model, which received FDA clearance in February 2025 as the first foundation model-powered clinical AI device. The CARE1 clearance included a PCCP, reflecting the reality that foundation models — which can be adapted to multiple clinical tasks through fine-tuning — require a regulatory framework that accommodates iterative improvement without requiring de novo review for each new application.
International harmonization is also advancing. In August 2025, the FDA, Health Canada, and the UK's Medicines and Healthcare products Regulatory Agency (MHRA) jointly published five guiding principles for PCCPs in machine learning-enabled medical devices. These principles cover:
- Transparency and documentation of planned changes
- Risk management throughout the device lifecycle
- Validation and performance monitoring
- Continuous improvement processes
- Stakeholder communication and labeling
For manufacturers, the decision to pursue a PCCP is strategic. Devices with predictable, well-characterized update cycles — such as radiology AI tools that require periodic retraining on new imaging data — are strong candidates. Devices with less predictable change patterns, or those where the impact of modifications is difficult to assess prospectively, may be better served by traditional submission pathways until the PCCP framework matures.
New Transparency and Labeling Requirements: Model Cards, Bias Evaluation, and AI Disclosure
The third pillar of the FDA's regulatory modernization effort addresses the transparency deficits that the JAMA study documented. The January 2025 TPLC draft guidance recommends that manufacturers provide structured, plain-language information about their AI devices through several mechanisms.
The most significant recommendation is the use of model cards — structured reports that describe an AI model's characteristics, intended use, performance metrics, limitations, and known biases. Model cards are not new to the AI industry — Google pioneered the concept in 2019 — but their incorporation into FDA guidance represents a formal regulatory endorsement. The guidance recommends that model cards include:
- Model input and output specifications
- Performance measures across relevant clinical subgroups
- Known sources of bias and their potential impact
- Demographic subgroup testing results (age, sex, race, ethnicity)
- Limitations and contraindications
- PCCP monitoring plans where applicable
The FDA also expects manufacturers to provide plain-language AI disclosure statements that are understandable and accessible to the intended users of the device. This goes beyond simply stating that a device uses AI — it requires explaining what the AI does, how it was validated, and what its limitations are in terms that clinicians and patients can act on.
| Transparency Requirement | What Manufacturers Must Provide | Regulatory Basis |
|---|---|---|
| AI Disclosure Statement | Plain-language description of AI functionality, validation approach, and limitations | TPLC draft guidance (Jan 2025) |
| Model Card | Structured report of model characteristics, performance, bias sources, and subgroup testing | TPLC draft guidance (Jan 2025) |
| Bias Evaluation Documentation | Testing results across demographic subgroups; data representativeness analysis | TPLC draft guidance (Jan 2025) |
| PCCP Monitoring Plan | Pre-specified performance metrics and acceptance criteria for planned modifications | PCCP final guidance (Dec 2024) |
| Post-Market Performance Data | Real-world performance tracking results and data drift analysis | TPLC draft guidance; Sept 2025 RFI |
Bias evaluation is a particularly demanding requirement. The guidance recommends that manufacturers test device performance across demographic subgroups and document any performance disparities. This is not a one-time exercise — the expectation is that bias monitoring continues throughout the device lifecycle, particularly when models are retrained or deployed in new populations.
Post-Market Monitoring Expectations: Data Drift and Real-World Performance
The FDA's post-market monitoring expectations for AI devices represent a significant departure from traditional medical device surveillance. Unlike a physical implant or a diagnostic reagent, an AI model's performance can degrade over time without any physical change to the device — a phenomenon known as data drift or model drift. Changes in patient populations, imaging equipment, clinical workflows, or documentation practices can all cause a model's performance to shift, sometimes dramatically.
The TPLC draft guidance addresses this by recommending that manufacturers establish ongoing device performance monitoring plans that include:
- Pre-defined performance metrics and thresholds that trigger investigation when breached
- Processes for detecting and quantifying data drift in input data distributions
- Mechanisms for collecting and analyzing real-world performance data
- Protocols for communicating performance changes to users and, where appropriate, to the FDA
In September 2025, the FDA issued a Request for Public Comment seeking evidence-based methods for measuring real-world AI device performance. This RFI signals that the agency recognizes the current state of post-market surveillance for AI devices is inadequate and is actively seeking input on how to improve it. The RFI covers topics including:
- Methods for collecting real-world performance data without imposing excessive burden on clinical sites
- Statistical approaches for detecting performance degradation in deployed AI models
- Standards for reporting real-world evidence to the FDA and to the clinical community
- Methods for evaluating device performance across diverse patient populations in real-world settings
The post-market monitoring expectations create practical infrastructure requirements for manufacturers. Teams need systems for collecting and analyzing performance data from deployed devices, processes for distinguishing between expected performance variation and genuine degradation, and clear escalation pathways when performance thresholds are breached. For manufacturers with devices deployed across multiple sites, this requires investment in data aggregation and analytics infrastructure that many do not currently have.
Practical Compliance Roadmap for Manufacturers in 2026
For regulatory affairs and quality assurance teams, the transition from the pre-market review paradigm to lifecycle oversight is not theoretical — it creates concrete actions that should be underway now. The following roadmap is organized by priority and timeline.
| Priority | Action | Timeline | Key Consideration |
|---|---|---|---|
| High | Audit existing device documentation against the nine TPLC areas | Q1–Q2 2026 | Identify gaps in device description, validation, and performance monitoring documentation |
| High | Evaluate whether planned algorithm updates qualify for PCCP submission | Q1–Q2 2026 | Focus on devices with predictable, well-characterized update cycles |
| High | Prepare model cards and bias evaluation documentation for active submissions | Q1–Q2 2026 | Include demographic subgroup testing results; document data representativeness |
| Medium | Establish post-market monitoring infrastructure | Q2–Q3 2026 | Define performance metrics, data drift detection methods, and escalation protocols |
| Medium | Review labeling for AI disclosure compliance | Q2–Q3 2026 | Ensure plain-language AI descriptions are accurate and accessible |
| Low | Monitor international harmonization developments | Ongoing | Track FDA-Health Canada-MHRA convergence; prepare for potential alignment |
The most urgent action is the documentation audit. Many manufacturers have device descriptions and validation documentation that were prepared under earlier FDA expectations. The TPLC guidance makes clear that the FDA now expects comprehensive documentation across all nine areas. Gaps in areas like data management, model description, and device performance monitoring are particularly common and will require significant effort to address.
The PCCP evaluation is equally urgent but more strategic. Not every device is a good candidate for a PCCP. The framework works best for devices where the manufacturer can clearly describe the types of changes they intend to make and can pre-specify the validation protocols for those changes. Devices with less predictable change patterns, or where the clinical impact of changes is difficult to assess prospectively, may be better served by traditional submission pathways for now.
Model card preparation should begin immediately, even for devices that are not currently under active submission. The model card format is becoming a de facto industry standard, and having one ready demonstrates regulatory maturity. The bias evaluation component is particularly important — manufacturers should be able to show that they have tested their device across relevant demographic subgroups and can document any performance disparities.
Comparison with the EU AI Act: Converging but Distinct Frameworks
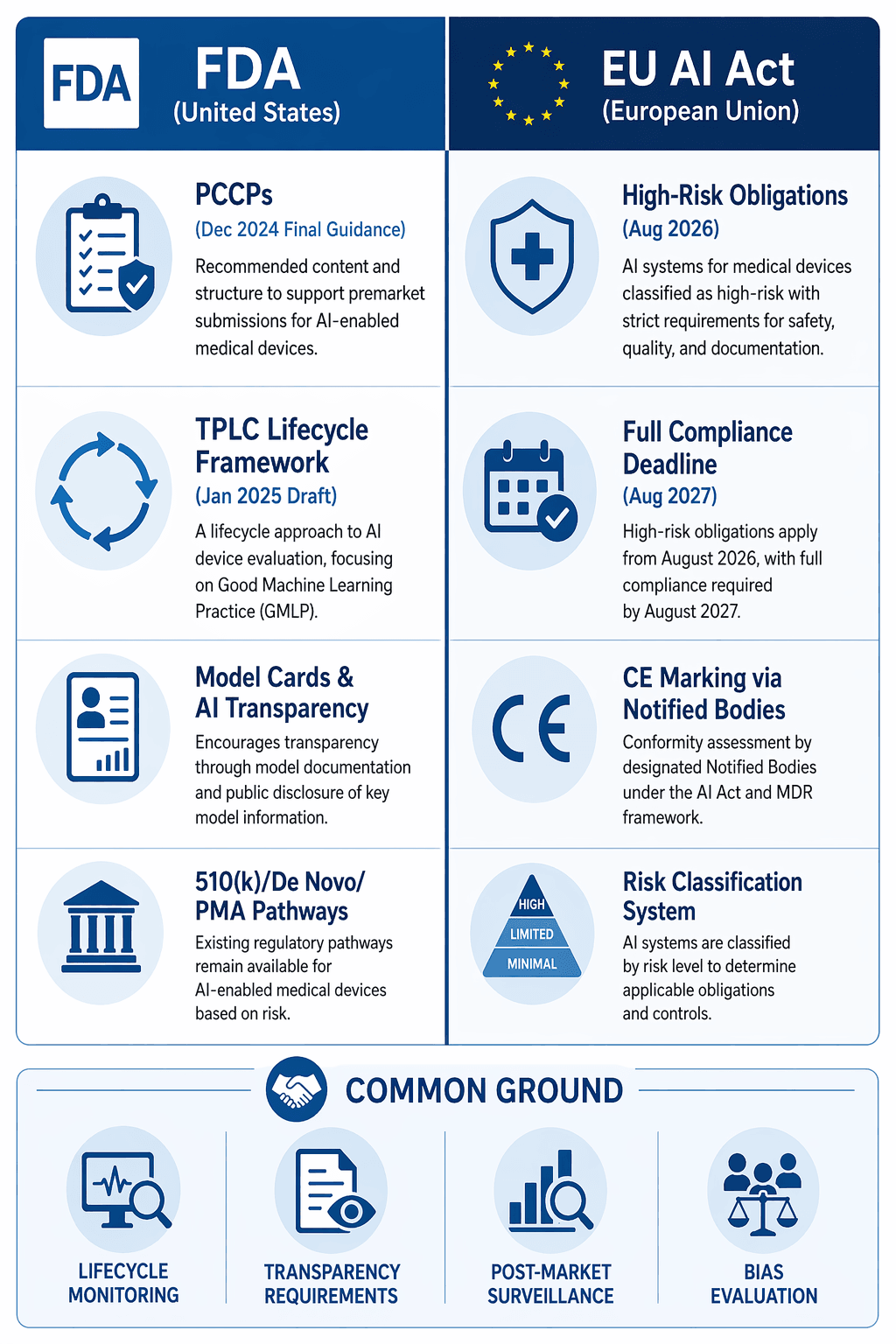
Manufacturers seeking to market AI-enabled medical devices in both the United States and the European Union must navigate two distinct but increasingly convergent regulatory frameworks. The EU AI Act, which entered into force in August 2024, classifies AI systems that are safety components of medical devices — or devices themselves — as high-risk. This classification applies to devices classified under MDR as class IIa, IIb, or III, and under IVDR as class A through D.
The timeline for compliance is accelerating. Most high-risk obligations under the AI Act take effect in August 2026, with full compliance required by August 2027. This means manufacturers have roughly 12 to 24 months to bring their AI-specific quality management systems into alignment with the Act's requirements.
| Dimension | FDA (United States) | EU AI Act (European Union) |
|---|---|---|
| Risk Classification | Device-specific (510(k), De Novo, PMA) | High-risk classification for MDR class IIa–III and IVDR class A–D devices |
| Lifecycle Approach | TPLC framework (draft guidance Jan 2025) | Lifecycle monitoring required under AI Act |
| Change Management | PCCP framework (final guidance Dec 2024) | Notified body involvement for substantial modifications |
| Transparency | Model cards, AI disclosure, bias documentation | Transparency obligations, human oversight, record-keeping |
| Post-Market Surveillance | Performance monitoring, data drift detection, Sept 2025 RFI | Post-market surveillance under MDR; AI Act record-keeping |
| Bias Evaluation | Demographic subgroup testing recommended | Data quality and governance requirements; bias mitigation expected |
| Conformity Assessment | FDA review (510(k), De Novo, PMA) | Single conformity assessment possible where notified bodies are accredited under both MDR and AI Act |
| Key Effective Date | PCCP final Dec 2024; TPLC draft Jan 2025 | High-risk obligations Aug 2026; full compliance Aug 2027 |
The common ground between the two frameworks is substantial. Both require lifecycle monitoring, transparency in how AI systems function and are validated, post-market surveillance, and bias evaluation. The EU AI Act layers AI-specific requirements — including data quality governance, human oversight, and record-keeping — on top of existing MDR/IVDR obligations. The Medical Device Coordination Group's MDCG 2025-6 guidance, published in June 2025, clarifies the interplay between MDR and the AI Act, noting that a single conformity assessment is possible where notified bodies are accredited under both frameworks.
The key differences lie in risk classification approach and conformity assessment pathways. The FDA's device-specific classification system (510(k), De Novo, PMA) differs fundamentally from the AI Act's high-risk classification, which applies automatically to certain device categories. Manufacturers should not assume that a device classified as low-risk under one framework will be similarly classified under the other.
For manufacturers operating in both markets, the most efficient approach is to build a quality management system that satisfies the most demanding requirements of each framework. The areas of convergence — lifecycle monitoring, transparency, post-market surveillance, and bias evaluation — should be treated as baseline requirements. The areas of divergence — particularly around risk classification and change management — require jurisdiction-specific planning.
The FDA's regulatory modernization through PCCPs, the TPLC framework, and transparency requirements represents a fundamental shift in how AI medical devices are regulated. For manufacturers, the message is clear: the era of minimal-evidence 510(k) submissions for AI devices is ending. The agencies are converging on a lifecycle oversight model that demands comprehensive documentation, ongoing performance monitoring, and proactive bias management. Manufacturers that invest in these capabilities now will be better positioned for both FDA and EU AI Act compliance in the years ahead.
Comments
Join the discussion with an anonymous comment.