The practical question is usually not, “How many AI devices has FDA authorized?” It is, “I have a clinical problem, a modality, or a workflow in mind; what has actually been authorized, and what is the device allowed to do?” That is where the search for FDA-authorized AI devices becomes both useful and easy to misread. The public FDA list is the right starting point, but it is not a polished clinical marketplace, an evidence database, or a complete registry.
The scale explains the friction. Public counts put AI/ML-enabled medical device authorizations at 1,451 through the end of 2025, including 295 in 2025 alone.[1] For more detail on that annual surge, see Did the FDA Authorize More AI Devices in 2025 Than Ever Before?. The official FDA page, meanwhile, presents its AI-enabled medical device list through downloadable table formats and links to individual authorization information; FDA also states that the list is not comprehensive because it is assembled by identifying AI-related terminology in publicly available summaries.[2]

Start With the FDA List, but Know What Its Fields Can and Cannot Tell You
The FDA list is still the source of record for the basic authorization trail. Its structured fields are administrative rather than clinical: date, submission number, device name, company, panel, and product code.[2] Those fields are exactly what a procurement analyst or regulatory reviewer needs to confirm that a candidate device exists in FDA’s public authorization materials. They are less helpful when the starting point is, for example, “stroke triage on CT angiography,” “diabetic retinopathy screening,” or “cardiac rhythm signal analysis.”
| FDA field | What it is good for | What it does not answer by itself |
|---|---|---|
| Date | Sorting authorizations chronologically and checking whether a device is recent | Whether the device is clinically better than older alternatives |
| Submission number | Finding the public FDA record and matching a device to a clearance or authorization pathway | Whether the study evidence is strong, representative, or prospective |
| Device name and company | Identifying the product and sponsor for follow-up | Whether the current marketed version matches the authorized description |
| Panel | Narrowing to a broad clinical area such as radiology or cardiovascular | Whether the device solves the specific clinical task being searched |
| Product code | Grouping devices that FDA classifies together | Whether all devices under that code use AI for the same function |
The product code deserves special caution. It is a useful regulatory handle, and it often gets a searcher closer to the right neighborhood. But it is not a reliable substitute for intended-use review. In an analysis of 1,016 FDA-authorized AI medical device authorizations, Singh and colleagues found that 19 of 69 multi-device product codes contained non-uniform AI-function taxonomies, and those product codes accounted for 57% of devices in the sample.[3] In plain terms: a product code can gather devices that do different AI jobs.
A Search Workflow That Holds Up Under Review
A defensible search does not need to be complicated, but it does need to keep discovery separate from verification. The fastest route is usually a loop: constrain the universe with the official list, broaden recall with semantic search, then return to the FDA record before making any claim about authorization or intended use.
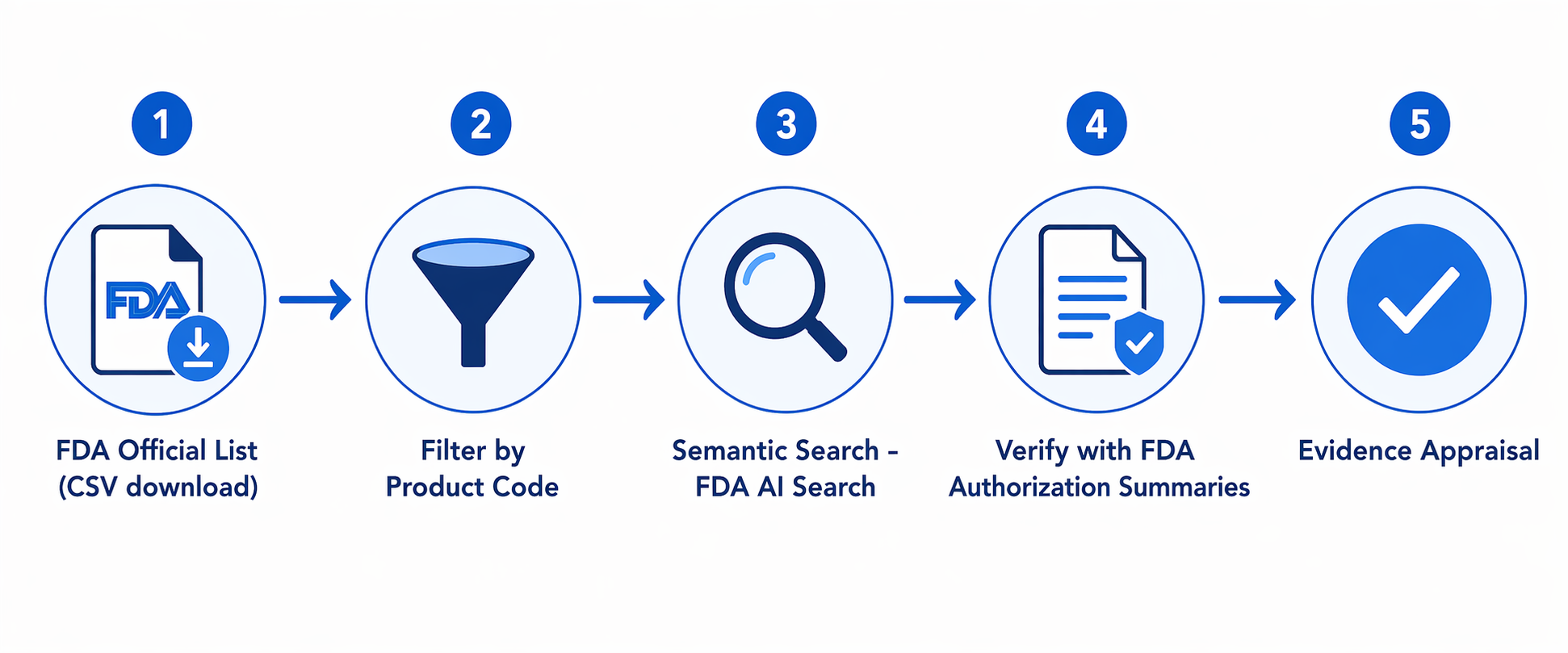
- Download the FDA AI-enabled medical device list and preserve a dated copy for your search file.
- Filter by panel, company, product code, date, or device name when you already know the regulatory neighborhood.
- Use the FDA product classification database or product-code lookup to understand what a code generally covers.
- Use FDA AI Search or another semantic layer when the query is clinical: disease, modality, body site, workflow, or intended task.
- Return to FDA summaries and other evidence sources before interpreting the result as suitable for procurement or clinical deployment.
When CSV Filtering Is Enough
CSV filtering is the cleanest first move when the search request already contains a known anchor. If a committee asks for AI devices from a specific manufacturer, for devices cleared in a particular year, or for all entries under a known FDA panel, the table can answer that quickly. It also gives you a reproducible trail: the downloaded file, filter criteria, and resulting rows can be saved and re-run when the list changes.
This matters because the FDA list is periodically updated and because public counts differ across analyses. The Imaging Wire reported 1,451 authorizations through the end of 2025, while other snapshots may show different totals depending on synchronization timing and how AI-related terminology is captured.[1][2] A dated CSV does not solve that discrepancy, but it prevents a later reviewer from wondering which version of the public list you searched.
When Product Codes Help—and When They Mislead
Product codes are helpful when the clinical question maps neatly to a device class. They can keep a search from drifting into unrelated specialties, and they are useful for finding predicate relationships or neighboring devices. If a cardiology team already knows the relevant code for a class of signal-analysis devices, product-code filtering can reduce a large table to a manageable set.
The mistake is to treat the code as if it describes the AI function. Singh and colleagues’ finding on non-uniform AI functions inside multi-device product codes is the reason product-code search should be treated as a constraint, not an answer.[3] After filtering by code, read the intended use and device description. Two devices that sit near each other administratively may not support the same workflow, input type, or clinical decision.
When Semantic Search Earns Its Keep
Semantic search becomes useful when the query is how clinicians actually speak: disease plus modality, image type plus task, or workflow plus setting. The official table does not naturally answer “AI for pulmonary embolism on CT,” “triage tool for intracranial hemorrhage,” or “retinal screening in primary care.” A keyword match may miss relevant entries if the public summary uses different phrasing.
FDA AI Search was built to address that gap. It is a third-party academic tool from Lotter Lab at Dana-Farber Cancer Institute, not an FDA product. Its paper describes a pipeline that extracts structured features with large language models, including keywords, thesis-like summaries, relevant questions, and query matches; embeds those features with MedEmbed; and combines embedding-based similarity across seven features with BM25 keyword scoring.[4]
On disease-plus-modality test queries, the reported hybrid approach achieved 82.9% Hit@1 and 95.1% Hit@3.[4] Those are search-performance metrics. They suggest the tool can surface relevant candidates more efficiently than table browsing for certain query types. They do not show that the returned devices improve outcomes, generalize to a local population, or fit a hospital’s workflow.
The safest way to use it is as a discovery layer. Enter the clinical phrase, collect plausible candidates, then verify each device name, sponsor, submission number, and intended-use language against FDA materials. If the semantic result and the FDA summary appear to disagree, the FDA source gets priority, and the discrepancy should be documented rather than smoothed over.
Radiology Searches Will Feel Different From Everything Else
Search expectations should change by specialty. Radiology dominates the FDA-authorized AI device landscape: The Imaging Wire reported that 76% of devices are radiology devices.[1] Singh and colleagues also found that 84.4% of devices in their analyzed sample used images as input, 14.5% used signals, and only 0.4% used tabular EHR data.[3]
That distribution has a practical consequence. A radiology procurement search may return a crowded field with multiple companies, adjacent indications, and overlapping modalities. A non-radiology search may return a short list or no obvious matches, not necessarily because the search was poor, but because the authorization landscape is uneven. For a deeper use-case view of the imaging market, see AI Medical Imaging Companies in 2026: A Use-Case-Driven Market Landscape.
The current list also should not be read as evidence that every AI category has reached market authorization. As of mid-2026, FDA had not authorized LLM-based medical devices, while Aidoc’s CARE1, cleared in February 2025, has been described as the first foundation-model-powered clinical AI device; FDA has indicated plans to tag such devices in future list updates.[2][3]
Finding a Device Is Not the Same as Understanding the Evidence
The most important limitation is not the inconvenience of the interface. It is what public authorization summaries may omit. Lin and colleagues examined FDA authorization summaries for 691 AI/ML devices through July 2023 and found that 46.7% did not report study design, 95.5% omitted demographic characteristics, and only 1.6% cited randomized clinical trial evidence.[5]
Those findings do not mean the devices are ineffective. They mean that a public summary may not contain enough information to judge effectiveness, fairness, deployment risk, or local fit. A procurement team that finds a relevant device still needs to ask what population was studied, what comparator was used, whether the device was evaluated prospectively, how performance changed across sites or subgroups, and whether the marketed version matches the authorized description.
This is also where authorization language matters. FDA authorization answers a regulatory question about a device for a stated intended use. It is not a blanket endorsement for every adjacent workflow a hospital might imagine. A tool cleared for triage, prioritization, detection, or quantification should be evaluated in that role, not quietly promoted into a broader clinical decision function.
For readers tracking how FDA is responding to evidence, lifecycle oversight, and transparency concerns, From Evidence Gaps to Lifecycle Oversight: How the FDA is Reshaping AI Medical Device Regulation covers the regulatory direction in more detail. Innolitics reported that predetermined change control plans appeared in 10% of 2025 AI/ML 510(k) clearances and that the median clearance time was 142 days, but those operational measures should not be confused with complete postmarket evidence.[6]
A Practical Search Pattern for a Real Request
Suppose a service line asks for AI software related to a disease and imaging modality. The first pass should not be a vendor demo or a general web search. Start with the FDA list and preserve the version searched. Filter to the likely panel if it is obvious. If the relevant product code is known, filter by it, but do not stop there.
Next, run the disease-plus-modality phrase through FDA AI Search or a comparable semantic layer. Add synonyms a clinician might use and terms that appear in radiology reports or workflow descriptions. Keep candidates that appear plausible, but record that the semantic tool is being used for discovery. It is allowed to be helpful without being authoritative.
Then go back to the FDA record for each candidate. Match the device name and company. Confirm the submission number. Read the intended use, indications, input type, user, and output. Note whether the device detects, flags, prioritizes, quantifies, segments, predicts, or supports another defined task. This is where many superficially similar results separate into different buckets.
Only after that should the evidence review begin. The FDA summary may provide useful performance information, but the Lin findings are a reminder to look beyond the summary when necessary.[5] Ask the sponsor for validation details, subgroup performance, implementation studies, cybersecurity and update practices, workflow integration evidence, and postmarket monitoring plans. If the request is for clinical use rather than market mapping, this step is not optional.
What Not to Conclude From a Search Result
- Do not conclude that the FDA list is exhaustive; FDA states that the list is not comprehensive.[2]
- Do not conclude that a product code means all devices under it perform the same AI function; product-code heterogeneity is documented.[3]
- Do not conclude that FDA AI Search validates a device; its reported metrics are about finding relevant records, not clinical performance.[4]
- Do not conclude that authorization summaries contain all evidence needed for procurement; key study and demographic details were often missing in Lin and colleagues’ sample.[5]
- Do not conclude that a device authorized for one workflow is appropriate for a neighboring workflow without checking the intended-use language.
The disciplined operating rule is simple: use third-party semantic search to find candidates, use FDA records to verify authorization and intended use, and use evidence appraisal beyond the authorization summary before procurement or clinical interpretation.
References
- Numbers from the FDA Show Radiology Is Maintaining Its Lead, The Imaging Wire, March 2026, link
- Artificial Intelligence-Enabled Medical Devices, FDA, link
- How AI is used in FDA-authorized medical devices: a taxonomy across 1,016 authorizations, npj Digital Medicine, 2025, link
- FDA AI Search: Making FDA-Authorized AI Devices Searchable, arXiv:2602.00006, 2025, link
- Benefit-Risk Reporting for FDA-Cleared AI-Enabled Medical Devices, JAMA Health Forum, September 2025, link
- 2025 Year in Review: AI/ML Medical Device 510(k) Clearances, Innolitics, link
Comments
Join the discussion with an anonymous comment.