A Pencil, Not a Knife
I accept the thesis that the FDA has operationalized lifecycle governance through binding guidance in 2025–2026. The four pillars are real. But I reject the implication that the simultaneous clinical decision support (CDS) deregulation is part of a coherent “dual-track” plan. The two moves serve different constituencies: the pillars impose cost on established SaMD manufacturers; the deregulation relieves startup and investor pressure. The line between high-risk and low-risk devices is drawn with a pencil, not a knife. And the evidence deficit means we cannot reliably distinguish tools that work from tools that merely look like they work.
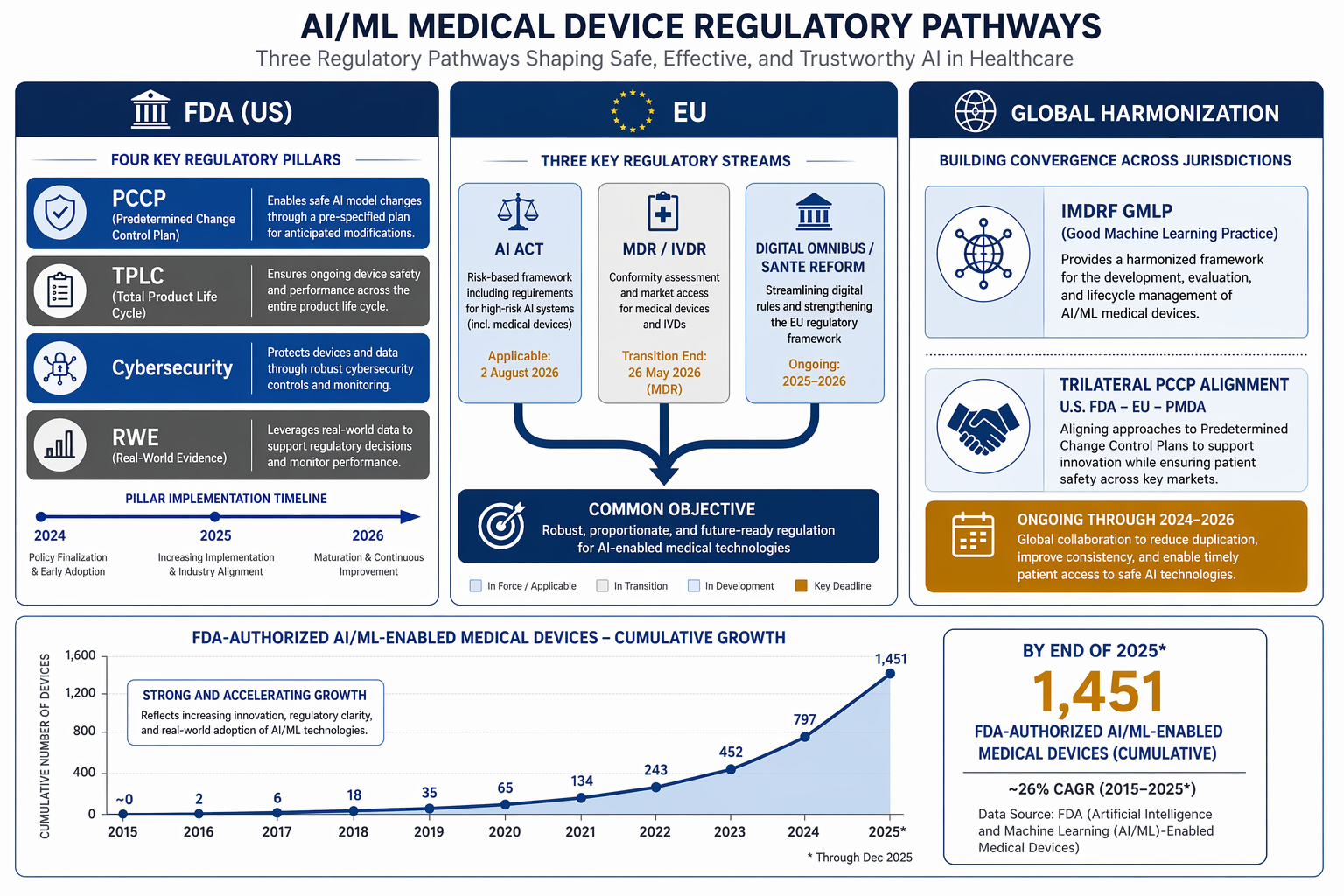
The FDA cleared 295 AI/ML-enabled medical devices in 2025 — a record. By year-end the cumulative count hit 1,451, according to the IntuitionLabs tracker. The number measures submission volume, not safety. What makes this period remarkable is the simultaneity of tightening and loosening.
The Four Pillars — and a Fifth
If you have been following FDA guidance cycles since the 2019 discussion paper, you know that “guidance” used to be a wish. The 2025–2026 pillars are not wishes — they are operational requirements with specific review gates and documentation mandates.
- PCCP (Final, August 2025): The Predetermined Change Control Plan lets manufacturers pre-authorize bounded algorithm modifications within the original submission. It defines boundaries, types of anticipated changes, and validation methodology. According to Proxima Clinical Research, a well-structured PCCP includes validation methodology and change types. This is not carte blanche to retrain on new data — the boundaries must be specified up front.
- TPLC (Draft, January 2025): The Total Product Lifecycle guidance lays out expectations for data lineage, bias analysis, human-AI workflows, transparency labeling, and post-market monitoring. As MD+DI reports, this is the first time the FDA has explicitly required lifecycle considerations in premarket submissions for AI devices.
- Cybersecurity (Final, June 2025): The updated guidance extends secure-by-design obligations to training data and model artifacts. Manufacturers must provide a Software Bill of Materials (SBOM) using CycloneDX or SPDX, along with threat modeling, penetration testing, and proof of a Secure Product Development Framework (SPDF). Proxima notes this documentation is now expected across all premarket pathways.
- RWE (Final, December 2025): The Real-World Evidence guidance expands the use of de-identified data for post-market monitoring. This opens the door for broader surveillance, but it does not mandate prospective studies.
QMSR alignment (effective February 2, 2026) is often treated as a separate item, but it is the fifth pillar for AI developers not already ISO 13485 certified. As Censinet explains, the rule aligns FDA quality system requirements with ISO 13485:2016. It affects documentation structure, not device performance, but it adds six to twelve months of preparation for a startup that was not already certified.
Comments
Join the discussion with an anonymous comment.