When a hospital reviews a cleared device using ML in healthcare, the first useful question is not whether the product appears on an FDA list. It is what kind of evidence sits behind that listing.
By the end of 2025, 1,451 AI/ML-enabled medical devices had been authorized in the United States, including 295 in 2025 alone. The scale is no longer marginal. Yet only 1.6% of FDA-cleared AI/ML devices cited a randomized clinical trial, and fewer than 1% reported patient health outcomes in FDA decision summaries.[1] That is the evidence problem clinicians inherit after market access has already been granted.

FDA authorization is meaningful. It determines whether a device can be legally marketed, and it reflects a regulatory review against the standard used for that submission. It is not, by itself, proof that a device improves outcomes in a particular hospital, patient population, service line, or workflow. That distinction is not semantics. It is the difference between accepting a product as legally cleared and deciding whether clinicians should let it influence patient care.
The public FDA list is still the right place to begin verification, and readers who need the mechanics can use a practical guide to the FDA AI-enabled medical devices database. But database presence should start an evidence review, not end it.
The Authorization Count Has Outrun the Outcomes File
The growth curve is easy to admire and difficult to govern. AI/ML device authorizations have expanded at about a 49% compound annual growth rate since 2016, with radiology accounting for roughly 76% of devices, cardiovascular applications about 9%, and neurology about 5%.[1] For a running view of the current clearance volume and specialty mix, ClinicalMind’s tracker of FDA AI device authorizations in 2025 is useful context.
Radiology’s dominance matters because many current devices are built around image detection, prioritization, triage, segmentation, or quantitative assistance. Those uses can be clinically valuable, especially where they reduce repetitive visual search. They also make the evidence landscape look more mature than it is. A model that detects a finding on an image may have good technical performance and still lack proof that its use changes diagnosis timing, treatment decisions, downstream testing, complications, length of stay, or mortality.
That is why the missing outcomes file matters. In decision summaries, fewer than 1% of cleared AI/ML devices reported patient health outcomes.[1] The absence of outcome reporting in a summary does not prove the manufacturer has no additional data. Some evidence may be unpublished, proprietary, or simply not captured in the public document. But for the clinician, quality officer, or imaging leader reviewing a device from the outside, public evidence is often the evidence available for institutional due diligence.
| Evidence Question | What The Public Record Often Shows | Why It Matters Locally |
|---|---|---|
| Was the device randomized in clinical use? | Only 1.6% of cleared AI/ML devices cited a randomized clinical trial.[1] | Randomization is one way to test whether use of the device changes care, not only whether the model performs on a dataset. |
| Were patient outcomes reported? | Fewer than 1% reported patient health outcomes in FDA decision summaries.[1] | A hospital may still need to test whether alerts, measurements, or prioritization affect actual patient management. |
| Are basic diagnostic metrics complete? | Only 29% of summaries included both sensitivity and specificity.[1] | One metric without its counterpart can hide the operational burden of false positives or false negatives. |
| Were race or ethnicity data disclosed? | Only 15.5% of 2024-authorized devices disclosed race or ethnicity data.[2] | Without subgroup transparency, local safety review has to account for uncertainty in the hospital’s own patient mix. |
| Can the algorithm change after clearance? | 16.7% of 2024 clearances included Predetermined Change Control Plans.[2] | Updateable models need monitoring after go-live, not just a one-time procurement review. |
Why 510(k) Clearance Creates This Gap
The structural hinge is the 510(k) pathway. About 97% of FDA AI/ML device entries relied on 510(k) clearance.[1] That pathway is built around substantial equivalence: the manufacturer shows that a new device is as safe and effective as a legally marketed predicate device for its intended use. It is not designed as a routine demand for new prospective clinical trials proving improved patient outcomes.
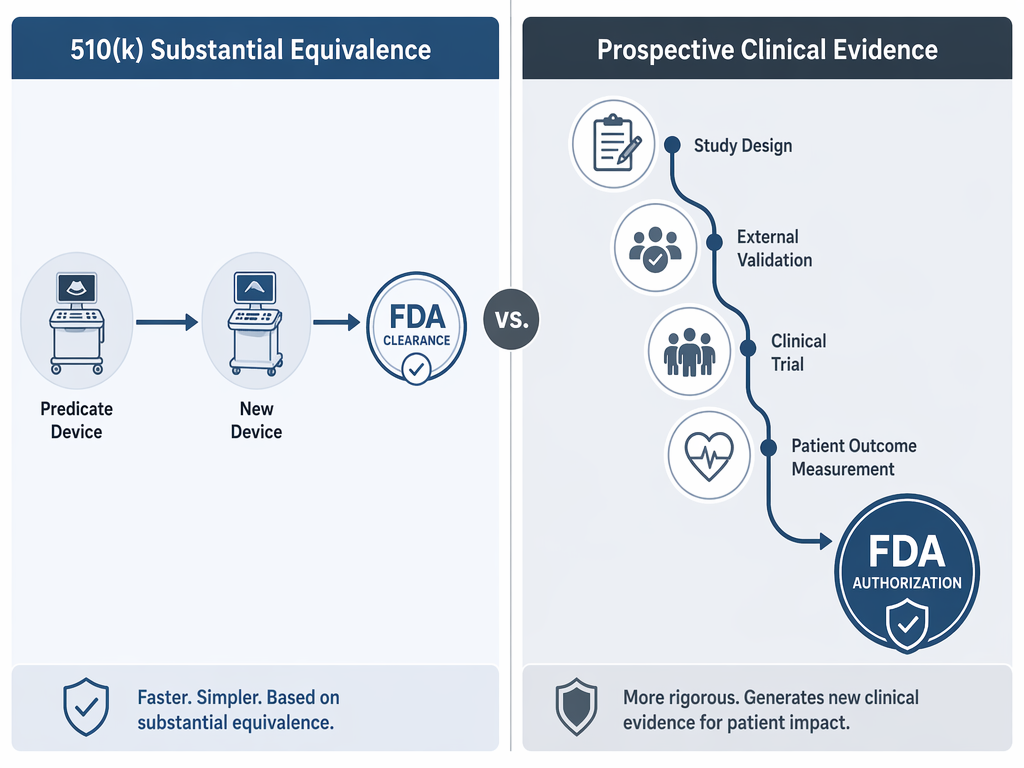
That distinction should lower the temperature of the debate. A 510(k) clearance is not a scandal simply because it is not a randomized trial. The pathway has a defined regulatory purpose. The problem starts when hospitals, vendors, or even clinicians allow “FDA-cleared” to carry more weight than the pathway was built to bear.
Substantial equivalence can be appropriate for market review while still leaving unanswered questions about clinical deployment. A device may be cleared for a narrow intended use, tested on retrospectively collected cases, and reported with model-level performance metrics. The hospital then has to decide whether the product should interrupt radiologist worklists, notify a stroke team, alter echocardiography review, flag incidental findings, or change who waits and who moves first.
Those are not the same questions. A regulator may ask whether a device meets the evidentiary threshold for the pathway. A health system must ask whether the device is safe and useful in its own patients, staffing model, imaging protocols, EHR environment, escalation rules, and quality metrics.
Performance Metrics Are Not the Same as Clinical Benefit
A familiar evidence packet for an AI/ML device often begins with sensitivity, specificity, area under the curve, accuracy, or agreement with expert readers. These measures are not trivial. A model that cannot detect the target condition reliably should not be excused by a pleasant user interface or a persuasive workflow story.
But the public record is inconsistent even at that level. Only 29% of FDA decision summaries included both sensitivity and specificity.[1] Reporting one without the other makes local interpretation harder. High sensitivity may come with a false-positive burden that changes worklist pressure, consult volume, alarm fatigue, or unnecessary downstream testing. High specificity may come with missed cases that matter most in time-sensitive pathways.
The deeper question is what the metric measures against. A model can perform well against an enriched test set and less well in routine care. It can perform well at one academic center and drift when used with different scanners, acquisition protocols, patient demographics, disease prevalence, or reporting conventions. It can flag the right image and still fail to improve care if the alert reaches the wrong person at the wrong time.
This is where procurement reviews often slide too quickly. Model validation answers whether the algorithm performed on the data used to test it. Clinical validation asks whether the device works safely and usefully when clinicians use it in care. Outcomes evaluation asks whether that use improves something patients or systems should care about. A cleared device may have evidence in one layer and very little in the others.
Demographic Opacity Is a Safety Issue, Not a Documentation Nuisance
The demographic reporting problem is especially hard to defend away. In 2024, only 15.5% of authorized AI/ML devices disclosed race or ethnicity data.[2] That figure does not prove that every undisclosed device performs inequitably. It does mean many public summaries do not give reviewers enough information to judge whether validation data resemble the patients who will be exposed to the device.
For some devices, subgroup performance may be clinically less central. For others, it is essential. Image quality, disease prevalence, comorbidities, access patterns, age distribution, device hardware, and site-of-care differences can all affect how an algorithm behaves once it leaves the development environment. If a product will be used across emergency, inpatient, outpatient, and community settings, demographic and site transparency become part of basic safety review.
A hospital does not need to reject every device with incomplete public subgroup reporting. It does need to treat missing subgroup evidence as a live implementation risk. That risk can be managed only if someone owns it: clinical leadership, quality, informatics, compliance, or a formal AI governance group. Otherwise, demographic uncertainty becomes invisible after contracting and reappears only when performance concerns emerge.
How to Read an Evidence Packet Before Go-Live
A useful review does not start from suspicion that every vendor claim is wrong. It starts from separating the claims by evidentiary burden. A tool that passively measures anatomy for clinician review carries a different burden than a tool that pages a team, reprioritizes a queue, or recommends an intervention. The more a device changes action, timing, or accountability, the more evidence it should carry.
Before local approval, reviewers should ask for the original FDA decision summary, the device’s intended use, the exact cleared indication, validation manuscripts if available, and any unpublished evidence the vendor expects the hospital to rely on. For search tactics, ClinicalMind’s guide to finding FDA-authorized AI devices efficiently is a practical starting point.
- Start with the claim: detection, triage, quantification, prediction, documentation support, or decision support. The evidence needed depends on what the device is actually allowed and expected to do.
- Identify the study design: retrospective dataset, reader study, external validation, prospective silent trial, prospective clinical deployment, or randomized clinical trial.
- Look for external validation: data from institutions, scanners, populations, and care settings outside the development environment.
- Separate model performance from workflow effect: a strong algorithm can fail operationally if alerts are ignored, delayed, duplicated, or routed to staff who cannot act.
- Demand subgroup reporting when the device will be used broadly: age, sex, race or ethnicity when available, site type, scanner or acquisition characteristics, and disease prevalence.
- Define post-market monitoring before deployment: false positives, false negatives, override rates, alert response time, downstream testing, clinician burden, and patient outcome measures when feasible.
The order matters. Many reviews jump from “FDA-cleared” to “Can IT integrate it?” That skips the clinical question. Integration is only worth solving after the organization knows which claim it is accepting and which residual uncertainty it is prepared to monitor.
A Simple Evidence-Grading Lens
| Evidence Level | What It Can Support | What It Does Not Prove |
|---|---|---|
| Bench or retrospective model testing | The model can perform a technical task on a defined dataset. | That use improves care in routine clinical workflow. |
| External validation | Performance generalizes beyond the original development setting. | Clinicians will respond appropriately or patients will benefit. |
| Prospective silent evaluation | The device can be tested against live local data without affecting care. | Clinical actions will improve once the device is activated. |
| Prospective clinical deployment study | The device can be assessed in real workflow with clinician behavior included. | Observed changes were caused by the device unless the design supports that inference. |
| Randomized clinical trial | The device’s effect can be compared against usual care under controlled conditions. | Performance will remain stable after scaling unless monitoring continues. |
This kind of lens prevents two common errors. One is treating clearance as if it were a full clinical evidence package. The other is treating anything short of a randomized trial as useless. Many low-risk tools can be adopted responsibly with careful validation, constrained use, and monitoring. Higher-risk tools need stronger evidence because their errors travel farther.
Vendor evaluation should also be tied to clinical function. A company selling worklist triage is making a different operational promise than one selling automated segmentation or ambient documentation. For a broader map of functions and companies, see ClinicalMind’s overview of healthcare AI companies by clinical function and evidence.
Radiology Is the Main Evidence Laboratory, But Not the Whole Story
Because roughly three-quarters of authorized AI/ML devices are in radiology, many institutional lessons are being learned there first.[1] Imaging departments have relatively structured data, defined acquisition systems, existing quality programs, and measurable workflow endpoints. That makes radiology a natural early market for AI/ML devices.
It also means the overall evidence picture is shaped by a narrow set of tasks. A clearance landscape dominated by image analysis does not automatically answer evidence questions for pathology, cardiology, neurology, monitoring, documentation, population health, or prediction tools. Readers focused on imaging can go deeper in ClinicalMind’s review of AI in medical image analysis.
The more a device moves away from bounded image interpretation and toward broader prediction or care coordination, the more careful the clinical validation question becomes. Predicting deterioration, prioritizing follow-up, or recommending action can entangle the model with staffing, access, baseline risk, and local treatment capacity. A technically accurate output can still fail if the system cannot respond.
PCCPs and Foundation Models Make the Review Less Static
Predetermined Change Control Plans are one sign that AI/ML regulation is adapting to software that may change after authorization. In 2024, 16.7% of AI/ML device clearances included PCCPs for post-market algorithm updates.[2] Properly used, a PCCP can define the types of planned modifications a manufacturer may make and the methods used to control those changes.
For hospitals, this adds a second review question. It is no longer enough to ask what evidence supported the device at clearance. Reviewers also need to ask what kinds of changes are anticipated, what performance testing will occur after updates, how users will be notified, and whether the institution can detect degradation in local use.
Foundation-model-powered systems complicate that further. Aidoc CARE1 received the first FDA clearance for a foundation-model-powered clinical AI device in February 2025.[3] That milestone does not by itself define the evidence standard for the category. It does show that reviewers should expect more devices whose internal development logic and update behavior do not resemble older, single-task software.
The practical implication is not to freeze adoption until regulation is perfect. It is to make change management part of clinical evidence review. If a model can change, the hospital’s assurance process has to continue after installation.
The Local Review Should Be Proportional to the Claim
A proportional review starts with clinical consequence. If a device produces an optional measurement that a specialist can easily verify, the review may focus on accuracy, usability, and documentation. If it changes triage, pages clinicians, suppresses cases, or influences treatment timing, the review needs workflow testing, safety monitoring, and a plan for missed or excessive alerts.
A reasonable local approval packet should answer these questions before routine use:
- What exact intended use did the FDA clear, and does the proposed hospital use match it?
- What comparator was used: expert readers, usual care, another device, adjudicated outcomes, or administrative labels?
- Were validation data external to the developer, and do they resemble the hospital’s patients and equipment?
- Are sensitivity and specificity both reported, and are thresholds fixed or adjustable?
- What are the likely false-positive and false-negative consequences in this workflow?
- Who receives the output, who is expected to act, and what happens when no one acts?
- What subgroup evidence is available, and what uncertainty remains for local patient populations?
- What post-market monitoring will continue after updates, staffing changes, scanner changes, or protocol changes?
This is also where hospitals should be careful with adoption narratives. Executive surveys and market reports can show interest, purchasing momentum, or perceived value. They do not replace evidence that a specific device improves a specific clinical process in a specific setting. For broader regulatory and adoption context, ClinicalMind’s companion piece on AI healthcare market regulation is the better place to weigh those pressures.
What Clearance Can and Cannot Do
FDA clearance remains necessary and important. It tells clinicians that the device passed a defined regulatory review for a stated intended use. It provides a legal and administrative boundary. It gives hospitals a starting record to examine.
It does not tell the whole clinical story. It may not show randomized evidence. It may not report patient outcomes. It may not provide complete diagnostic metrics. It may not disclose enough demographic detail for equity review. It may not answer whether a local workflow can absorb the device safely.
The disciplined position is neither blanket enthusiasm nor reflexive rejection. Cleared AI/ML devices may be useful, sometimes very useful. But the evidence demanded should be proportional to the clinical risk, workflow impact, patient population, and claim being made. A hospital that treats FDA clearance as the beginning of that review, rather than the conclusion, is asking the right question.
References
- JAMA Health Forum adoption study, JAMA Health Forum, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC12639477/
- Biomedicines 2025 analysis, Biomedicines, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC12730494/
- FDA AI Medical Device Tracker, IntuitionLabs, /clinical-applications/fda-ai-device-authorizations-2025
Comments
Join the discussion with an anonymous comment.